
Abstract
Epstein–Barr virus, a ubiquitous human herpesvirus, can induce both lytic and latent infections that result in a variety of human diseases, including lymphoproliferative disorders. The oncogenic potential of Epstein–Barr virus is related to its ability to infect and transform B lymphocytes into continuously proliferating lymphoblastoid cells. However, Epstein–Barr virus has also been implicated in the development of T/natural killer cell lymphoproliferative diseases. Epstein–Barr virus encodes a series of products that mimic several growth, transcription and anti-apoptotic factors, thus usurping control of pathways that regulate diverse homeostatic cellular functions and the microenvironment. However, the exact mechanism by which Epstein–Barr virus promotes oncogenesis and inflammatory lesion development remains unclear. Epstein–Barr virus-associated T/natural killer cell lymphoproliferative diseases often have overlapping clinical symptoms as well as histologic and immunophenotypic features because both lymphoid cell types derive from a common precursor. Accurate classification of Epstein–Barr virus-associated T/natural killer cell lymphoproliferative diseases is a prerequisite for appropriate clinical management. Currently, the treatment of most T/natural killer cell lymphoproliferative diseases is less than satisfactory. Novel and targeted therapies are strongly required to satisfy clinical demands. This review describes our current knowledge of the genetics, oncogenesis, biology, diagnosis and treatment of Epstein–Barr virus-associated T/natural killer cell lymphoproliferative diseases.
Similar content being viewed by others


Introduction
Epstein–Barr virus (EBV), a member of the human herpesvirus family, possesses oncogenic potential through its ability to infect and transform B lymphocytes into continuously proliferating lymphoblastoid cells. EBV infrequently infects T cells and natural killer (NK) cells and can lead to a wide range of T/NK cell lymphoproliferative diseases (LPD). It is conceivable that pre-existing inflammatory lesions, such as those caused by mucosal pathogens or inhaled materials that become lodged in the nasal mucosa, may induce local EBV-infected memory B cells to enter the lytic cycle and thereby transmit virus to locally activated T and/or NK cells. Persistent EBV infection is a risk factor for a wide range of human tumors and malignant diseases such as T/NK cell LPD.
Biological functions of T and NK cells and EBV infection
The T-cell compartment is divided into CD4+ and CD8+ T cells; these are referred to T helper and cytotoxic T cells, respectively. These cells play critical roles in the immune system and in the regulation of immune responses.1 NK cells initiate innate immune responses against invading pathogens and cancers.2 NK cells are characterized by the absence of T-cell receptor (TCR) gene rearrangement, lack of expression of the TCR-CD3 complex and the expression of CD16 and CD56.3 NK cells and cytotoxic T cells share a close relationship in terms of ontogeny and function.4
EBV infects a very broad spectrum of in vivo target cells, including B and T lymphocytes, NK cells, squamous and glandular epithelial cells, and smooth muscle cells.5 Although EBV infection is normally restricted to B lymphocytes, the virus has also been strongly linked to tumors of a T/NK cell origin after the aberrant virus has gained entry into T or NK cells. The intracellular signals within natural target cells that normally govern viral behavior may cease to function properly, allowing EBV to maintain a lifelong persistent latent infection in the host.6
EBV is transmitted primarily through infected saliva and establishes a latent infection in B lymphocytes in episomal (circular) DNA form by undergoing episodic lytic replication in B cells and epithelial cells, leading to viral reproduction and high levels of salivary shedding in the throat.7 The EBV life cycle demonstrates a number of distinctive viral features that are also typical of other gamma herpesviruses, as follows:6 (i) Lytic infection (primary infection) most likely occurs when EBV replicates in squamous epithelial cells and possibly locally infiltrating lymphocytes. (ii) EBV colonizes B cells via growth transformation in oropharyngeal lymphoid tissues. (iii) EBV downregulates growth-transforming gene expression in the transformed cells. (iv) In latent infection, EBV-infected but quiescent memory cells persist in the recirculating memory B-cell pool. (v) In some cases, latently infected B cells enter the lytic cycle; when this occurs at a mucosal surface, the shed virus particles can infect new host cells and produce new growth-transforming B-cell infections.
Exposure to EBV usually occurs early in childhood, and more than 90% of adults worldwide are EBV seropositive. Most primary EBV infections are asymptomatic in young children, although some cases may present as acute infectious mononucleosis if infection is delayed until the second decade of life or later.7, 8, 9 Persistent EBV infection is a risk factor for a wide range of human tumors.
During the EBV life cycle, some imbalances between the inherent transforming abilities of the virus and the host immune system can lead to the development of different diseases.10 These imbalances include suppression of the most immunogenic latent proteins, expression of lytic proteins that interfere with antigen-processing machinery and major histocompatibility complex molecule expression in infected cells, and production of viral homologues of human cytokines.10
In immune-competent hosts, protective immunity is mediated by strong cell-mediated responses to primary infection. These responses involve NK cells, CD4+ T cells, and particularly, EBV-specific cytotoxic CD8+ T lymphocytes (CTLs) and act collectively not only to control the primary infection and limit periodic reactivation but also to control the re-emergence of virus-transformed lymphoproliferative lesions.6 After primary infection clearance, EBV persists as a lifelong latent infection in infected memory B cells by suppressing the expression of the most immunogenic latent proteins and expressing only nonpathogenic and completely silent transcripts for EBV small RNAs (EBERs).11 Thereby, EBV can escape immune recognition and establish a true antigen-negative form of latency (L0) within cells in the recirculating memory B-cell pool.6 In immune-compromised hosts, however, primary EBV infections may efficiently transform B cells, resulting in life-threatening diseases.6
Oncogenesis of EBV-positive T/NK cell LPD
EBV gene expression and cellular genomic alterations
Like all herpesviruses, EBV experiences both latent and lytic replication stages, allowing it to evade immune surveillance and maintain lifelong infection. Latent infection is characterized by the absence of infectious virus production and integration into host cell chromosomes via replication of the viral genome, whereas lytic infection occurs when the virus produces a large number of functional and structural proteins to replicate its DNA and produce infectious viral particles.12, 13 After lytic replication, the expression of latent programs is required to trigger associated diseases.14 The EBV genome limits gene expression to nine latent viral proteins in varying patterns to evade immune recognition within infected resting memory cells. Latent proteins, including nuclear antigens (EBV-determined nuclear antigen (EBNA)-1, -2, -3A, -3B and −3C) and leader protein, are responsible for maintaining the viral genome as well as controlling the expression of three latent membrane proteins (LMP-1, -2a and -2b). BamHI A rightward transcripts (BARTs) and two small non-coding RNAs, EBER-1 and EBER-2, are also expressed (Table 1).15, 16 Latency can be classified into three patterns, depending on which latent genes are expressed.9 Overexpression of these oncoproteins is an important mechanism of associated lymphoproliferative disorders (Figure 1).

Epstein–Barr virus (EBV)-encoded proteins are associated with cellular proliferation, survival, differentiation and angiogenesis. Lytic cycle genes BCRF1 (viral IL-10) and BARF1 (sCSF-R) facilitate blunting of T-cell responses by suppressing antiviral cytokine production. BHRF1, a Bcl-2 homologue, preserves mitochondrial membrane potential and contributes to apoptosis resistance. Latent genes (EBNA1, EBNA2, LMP2A and LMP1) also protect host B cells from multiple apoptotic stimuli mediated by p53, Nur77, BCR and DR signals. For example, LMP1-mediated NF-κB activation upregulates several antiapoptotic genes capable of blocking intrinsic and extrinsic cell death pathways.
LMP-1, the main oncogenic protein of EBV, is considered an analogue of TNF-receptor 117 and provides both growth and differentiation signals to B cells. LMP-1 can activate several downstream signaling pathways that contribute to the induction of genes encoding anti-apoptotic proteins (for example, Bcl-2 and A20) and cytokines (for example, interleukin (IL)-10 and CD40L) and induces immortalization in B cells. Involved signaling pathways include the nuclear factor kappa B (NF-κB), MAPK/ERK1/2,13 PI3K/Akt, Notch, Jun N-terminal protein kinase (JNK),17 JAK/STAT, extracellular signal regulated kinase (ERK), interferon-regulatory factor 4 (IRF4) and Wnt pathways.14 In addition, the PI3K/Akt pathway seems to be most important in EBV-induced malignancies. This pathway is activated by LMP-1 in a manner that depends primarily on its C-terminal tail signaling domains, and in particular, the carboxy-terminal activating region 1 (CTAR1).17 Moreover, activation of the PI3K/Akt pathway and its downstream effector Bcl-2 will in turn suppress the pro-apoptotic activity of prostate apoptosis response-4.18 LMP-1 also downregulates BLIMP1α expression to prevent the differentiation of B cells to plasma cells, an important step related to lymphomagenesis (Figure 2).19

Signaling pathways in T/NK-cell proliferation. NF-κB pathway in EBV-positive T-cell lymphoma: a diagrammatic depiction of the pathogenesis and molecular mechanisms associated with progression from hemophagocytic lymphohistiocytosis (HLH) to chronic active disease or T-cell lymphoma in Epstein–Barr virus (EBV)-infected T cells. EBV latent membrane protein-1 (LMP-1) upregulates tumor necrosis factor-α (TNF-α) via TNF receptor (TNFR) associated factors (TRAF)/nuclear factor-κB (NF-κB) signals on one hand to kill bystander lymphoid cells and downregulates TNFR-1 on the other hand to suppress the apoptotic signaling pathway, thus conferring survival from TNF-α-induced apoptosis on LMP-1-expressing T cells.
LMP2 is transcribed from two different promoters to produce either LMP2A or LMP2B. It plays a key role in inhibiting normal B-cell development by suppressing B-cell receptor-mediated proliferation signals through inhibition of calcium mobilization and tyrosine phosphorylation and thus plays a key role in abrogating normal B-cell development and activation of the lytic viral replication cycle. LMP2 also provides the tonic signals required for B-cell survival by co-opting SYK and SRC-family kinases.20 In AIDS-related lymphomas, LMP2A also contributes to NF-κB signaling via LMP1 signaling activation.12
EBNA-1 is the only consistently expressed latent protein in all EBV-positive malignancies. It plays a key role in maintaining EBV in infected cells and facilitating episomal replication. EBNA-1 has also been characterized as a transcriptional activator and activates EBV transcript expression via the latent C promoter.8 EBNA2 plays an important role in B-cell transformation. It initiates and maintains transformed cell proliferation and prevents transformed B-cell apoptosis.8
EBERs are small noncoding RNAs that are expressed in EBV-infected cells and have been reported to protect cells from apoptosis through IRF-3 and NF-kB signaling.12, 20 EBERs can also suppress IFN-α-mediated apoptosis; induce growth-promoting cytokines such as IL-10, IGF-1, IL-9 and IL-6; induce B lymphocyte growth transformation;20 and upregulate the Bcl-2 oncoprotein. MicroRNAs are also expressed during EBV latency. In human primary B cells, microRNAs in the BHRF1 region inhibit apoptosis and contribute to cell cycle progression and proliferation during the early infection phase.21
The lytic cycle also contributes to the growth of EBV-associated malignancies by enhancing angiogenesis. B cells infected with a virus competent for expression of the lytic protein BZLF-1 release greater amounts of vascular endothelial growth factor and IL-6 than do cells infected with BZLF-1-defective virus.22 Lytically infected cells have been suggested to promote EBV-driven lymphomagenesis.23 BZLF1 is a master regulator of the expression of several early viral genes critical to productive viral replication and is sufficient alone to activate the entire lytic cascade.8
Immunologic mechanism
Primary immunodeficiencies
Some types of primary immunodeficiencies are well known because their main feature is the development of EBV-associated disease.9 These immunodeficiencies mainly comprise defects related to the lymphocyte cytotoxic pathway or T-cell dysfunction, including disruptive interactions between B cells and T cells. These genetic defects are responsible for the development of an acute fulminant life-threatening condition after EBV infection.7
Acquired immunosuppression
The manifestation of EBV-related tumors often varies according to the patient’s immune status (for example, HIV infection or transplant-related immunosuppression). In healthy individuals, the lifelong asymptomatic latency established by EBV in B lymphocytes is effectively controlled by EBV-specific CTLs after primary infection. In transplant patients, however, the administration of powerful immunosuppressive agents impairs CTL responses, thus allowing virus-infected B cells to accumulate and possibly leading to uncontrolled EBV-driven lymphoproliferation and tumor formation.24 The resolution of a high percentage of posttransplant lymphoproliferative disorders (PTLDs) in response to a reduction in immunosuppression as well as the success of donor lymphocyte infusion25 strongly suggests that the underlying state of immunosuppression is among the most important permissive factors for PTLD development.
Infiltration of regulatory T cells
EBV-positive malignant cells can attract infiltrating T regulatory cells in the tumor microenvironment or can induce differentiation of the Tr1 phenotype from naïve CD4+ T cells in the tumor lesion. At increasing numbers and cell activation levels, regulatory T cells can inhibit anti-tumor immunity in the EBV-associated cancer-bearing host that has maintained a long-term latent EBV infection. In addition, the number of infiltrating regulatory T cells and their activation status will affect tumor development and patient outcomes in cases of EBV-positive malignancies.26
Defects in lymphocyte cytotoxic function and NK cells
CD8+ T lymphocytes and NK cells are essential for immunosurveillance against cellular anomalies and virus-infected cell elimination. Defects in CTL and NK cell cytotoxic function preclude downregulation of the elicited immune response, resulting in persistent hyper-activation and proliferation of these effector cells.7 This condition also leads to an uncontrolled but ineffective immune response mediated through the granule-dependent pathway, resulting in hemophagocytic lymphohistiocytosis (HLH).27
Defects in T-cell signaling and T-cell/B-cell interaction
A heterogeneous complex of T-cell defects that may essentially preserve CTL function but exhibit genetic aberrations in intracellular T-cell signaling and/or T cell–B cell interactions can occur and affect T-cell survival, proliferation, differentiation, homeostasis and migration. These defects involve the signaling lymphocytic activation molecule (SLAM), SLAM-associated protein (SAP), X-linked inhibitor of apoptosis (XIAP), IL-2-inducible T cell kinase (ITK) (Figure 3), magnesium transporter 1 (MAGT1) and coronin-1A.7 The programmed death (PD)-1/PD-1 ligands (PD-Ls) pathway, a new member of the B7/CD28 family, is also involved in various T-cell-mediated diseases in reactive lymphoid tissues and inhibits tumor-associated T-cell activity.28

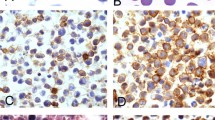
(A) IL-2-dependent tumor cell proliferation in Epstein–Barr virus (EBV)-positive NK-cell LPD. Activated tumor-infiltrating T cells produce inflammatory cytokines such as IL-2 and the related cytokine IL-15 (far left panel). In the next panel, these lymphocytes make contact with malignant cells and supply IL-2, which in turn induces IL-10. In the next panel, IL-10 elevates the level of LMP-1 in the EBV-infected cells, which consequently upregulates CD25 expression (IL-2R-α). Finally, a lower IL-2 concentration can greatly induce targeted cell proliferation after IL-2R upregulation. (B) Morphologic and pathologic presentations of NK/T-cell lymphoma subtypes. Extensive coagulative necrosis is observed (a). Tumor cells generally exhibit an angiocentric growth pattern (b). Pleomorphic large atypical cells, some of which feature a cucumber-like morphology (c). In situ hybridization for EBV-encoded early small RNA (EBER) shows numerous positive cells (d).
Chronic inflammation
Abnormal T lymphocyte cytotoxic activity fails to clear EBV-infected cells, resulting in the continuous activation and proliferation of both CTLs and NK cells. Various murine gene knockout models have formally demonstrated the involvement of pro-inflammatory cytokine genes during tumor development.29 Cytokine-induced mutagenesis is one such mechanism. Activated inflammatory cells induce reactive oxygen species-associated DNA damage and genomic instability. Mutagenesis may also repress mismatch repair response genes or induce the ectopic expression of activation-induced cytosine deaminase, which is normally involved in the somatic mutation of immunoglobulin genes in B cells but can cause off-target effects. Furthermore, cytokines released from inflammatory cells may mediate growth promotion by activating the NF-κB, STAT3 or AP1-associated growth or pro-survival pathways, leading to the proliferation of malignant cells and promotion of tumor angiogenesis.30
Clearly, chronic active EBV infection, HLH and T/NK-cell lymphomas are all associated with an inflammatory environment containing high serum pro-inflammatory cytokine levels. Factors related to EBV-positive T/NK-cell growth include IL-2 (Figure 2), IL-9, IL-10, CD70 and sCD27.6 Invasive factors such as IFN-γ, IP10 and IL-15 contribute to tumor infiltration.
Age-related immunosenescence
Numerous factors and complex mechanisms, such as telomere shortening, are involved in immune system remodeling during the aging process. The activation of telomerase, which is critically involved in telomere length maintenance, is also required for the transformation of virus-infected primary B lymphocytes; these cells are critically involved in maintaining telomere length and overcoming replicative senescence to facilitate unlimited replication.31 LMP-1 was found to activate transcriptase (TERT) at a transcriptional level via the NF-κB and MAPK/ERK1/2 pathways.32, 33 Other factors that contribute to lymphomagenesis include alterations in T-cell homeostasis and transduction, impaired DNA repair and dysregulated antioxidant mechanisms.13
Other cofactors
Increasing evidence has shown that cofactors may play a role in the development of EBV-associated neoplasms. These cofactors include genetic susceptibility, environmental factors, host immune status reactivation and nutritional status.34
Genetic susceptibility has been identified in several diseases associated with EBV infections. A clear association exists between EBV and extranodal NK/T-cell lymphoma, nasal type. This malignancy, of which each case is EBV-genome-positive, is characterized by vascular damage, necrosis and a cytotoxic T-cell phenotype.12 It is most common in Asia, followed by Native Latin America where the populations are somewhat genetically linked with Asian populations.35 People in these areas may harbor genetic susceptibility. Regarding nasopharyngeal carcinoma, a surface epithelial tumor, a genetic susceptibility in some individuals (particularly human leukocyte antigen haplotypes) is a well-defined etiological factor.36
Environmental factors such as parasitic infections may also play an important role in EBV-positive T/NK-cell LPD, similar to the roles played by malaria and EBV as co-factors in Burkitt lymphomagenesis and by HIV and EBV as co-factors in B-lymphomagenesis.6 Regarding gastric carcinoma, the status of EBV as a co-factor versus Helicobacter pylori is key and cannot be ignored. The possibility of synergy between these two infectious agents has been suggested by a recent study of gastritis in pediatric patients, in which individuals with serologic evidence of Helicobacter pylori and EBV co-infection were more likely to develop more severe inflammatory lesions than were those with Helicobacter pylori infection alone.37
Additionally, the host immune status is essentially related to disease development. As previously mentioned, impaired immunosurveillance against EBV may favor the development of EBV-associated diseases in posttransplant patients and HIV-1-infected individuals. For example, patients with AIDS-associated leiomyosarcoma are all EBV-genome-positive, an incidence that is much higher than that among HIV-negative patients.38
The host nutritional status is also an important factor. Malnutrition and the consumption of food with possible carcinogens such as volatile nitrosamines may also contribute to the development of these diseases.
Genetics of EBV-positive T/NK-cell LPD
Cellular genetic alterations are of great importance to pathogenesis. Such alterations include gene deletion, gene rearrangement, gene insertion and tumor-suppressor gene mutation, and are critical for proliferation, apoptosis and differentiation in lymphomagenesis. Although most of these mechanisms occur mainly in B-lymphomagenesis, similar effects may also be relevant in T/NK-cell infection.
In T/NK-cell infection, the risk of genome mutation increases in latency I/II infection (expression of EBNA1 and LMP1). EBNA1 activates reactive oxygen species production that induces chromosomal aberrations and double-strand breaks, whereas LMP1 promotes genomic instability by inhibiting DNA repair pathways and suppressing the DNA damage checkpoint.39, 40 Studies of nasal tumors have found chromosome 6q21-25 deletions, CD95 (Fas) gene mutation and TP53 gene mutation in some cases.41 Loss of chromosome regions at 9p21 and 3p is also commonly observed and thought to occur during early nasopharyngeal carcinoma pathogenesis. The high frequencies of 3p and 9q losses may contribute to latent EBV infection, a crucial event in the multistep progression towards nasopharyngeal carcinoma.42, 43
MYC is a nuclear phosphoprotein with gene-activating and -repressing capabilities. Lymphomas expressing latency I exhibit cellular oncogenic alterations such as translocations involving the MYC oncogene, which are characteristic of Burkitt lymphoma.44 Therefore, it is likely that EBNA1 protects against apoptosis by dysregulating the expression of MYC, which would be further enhanced by expression of the Bcl-2 homologue BHRF1.45 Other types of LPDs with type I EBV latency exhibit additional transforming alterations.12 In the nasal type, genetic alterations include an absence of T-cell antigens, expression of the NK cell marker CD56 and absence of TCR gene rearrangement.46
Tumor-suppressor gene mutation, including TP53 mutation, has been observed in numerous cases.6 In PTLD, different types of molecular gene alterations have been recognized, including microsatellite instability, altered proto-oncogene (MYC) or suppressor gene (Bcl-6 and TP53) functionality, DNA hypermethylation and aberrant somatic hypermutation.47 Similar to nasopharyngeal carcinoma, EBV-associated gastric carcinoma tumors display a type II latency EBV latent gene expression program. Compared with EBV-negative GC, EBV-associated gastric carcinomas have distinct phenotypic and clinical characteristics, including absent p16 expression, p73 promoter methylation, wild-type TP53 expression, different allelic loss patterns and improved patient survival.36
Additionally, gene insertion was observed in EBV isolates from malignant tumors.48 Rb gene mutation has been suggested as a pathogenic mechanism in some T-cell lymphomas.49 Inactivation of the CDKN2 genes, which encode the p16INK4a and p14 (ARF) proteins, occurs in the majority of human T-cell acute lymphoblastic leukemias.50, 51 Hypermethylation of the cyclin-dependent kinase inhibitor (CDKI) gene, p15INK4B, may also be involved in the pathogenesis of T-cell acute lymphoblastic leukemias.52 The EBV nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression.53 Induction of p16INK4a is the major barrier to proliferation upon the EBV-mediated transformation of primary B cells into lymphoblastoid cells.54
Diagnosis of EBV-positive T/NK-cell LPD
T-cell lymphoma
Peripheral T-cell lymphoma, unspecified
Peripheral T-cell lymphoma, unspecified (PTCL-U) typically occurs in adults (median age, 60 years) with a higher prevalence in men.55 The majority of cases are nodal in origin. However, extranodal involvement is also common and most often involves the skin and gastrointestinal tract. Bone marrow involvement can occur in 20–30% of cases. Eosinophilia, thrombocytopenia and elevated LDH are common, as well as pruritus and hemophagocytic syndromes.56 Systemic constitutional symptoms (B symptoms: fever, weight loss, night sweats) are common. Approximately 65% of patients have stage IV disease and 50–70% of patients are in the high or high-intermediate group according to International Prognostic Index scoring.57, 58 The morphology of PTCL-U is highly variable. PTCL-U, which is characterized by an inflammatory background, typically exhibits paracortical or diffuse infiltrates with a mixture of small and large cells effacing the lymph node architecture. The cytological spectrum is very broad, ranging from highly polymorphous to monomorphous. Clear cells, follicular dendritic cells, eosinophils and Reed–Sternberg-like cells might also be observed. High endothelial venules are often increased.59, 60 Immunohistochemistry is important for a diagnosis of PTCL-U. PTCL-U is usually characterized by CD3 expression. CD7 expression is most commonly absent, whereas CD5 and/or CD2 positivity are frequent.59 Most nodal cases express CD4 and lack CD8. However, CD4/CD8 double-positivity or double-negativity may be observed. CD8, CD56 and cytotoxic granule expression are occasionally positive. PTCL-U exhibits CD52 positivity but usually lacks CD10, Bcl-6, PD1 and CXCL13.61, 62, 63, 64 In more than 50% of cases, the integration of EBV, which is present in bystander B cells and/or a variable fraction of the tumor cells, has been reported and is considered to be a risk factor for survival.65, 66
Angioimmunoblastic T-cell lymphoma
Angioimmunoblastic T-cell lymphoma (AITL) is a rare neoplasm but one of the most common peripheral T-cell lymphoma subtypes.57, 67 AITL mostly occurs in older adults (median age, 59–65 years), with a slightly higher prevalence in men.57, 67 Patients often have B symptoms, generalized lymphadenopathy and hepatosplenomegaly. Bone marrow involvement is also frequently observed.68, 69
AITL lesions exhibit a polymorphous infiltrate containing atypical medium-sized neoplastic cells with clear cytoplasm and prominent arborizing blood vessels that are admixed with small lymphocytes, histiocytes, immunoblasts, eosinophils, plasma cells, increased follicular dendritic cells and scattered EBV B-cell blasts.62, 70, 71, 72 EBV can be detected only in B cells. In some AITL patients, EBV likely plays a role in the development of EBV-associated B-cell lymphoma. In addition to EBV, HHV6B, another human herpesvirus, has also been reported in approximately half of AITL cases.73
Mature CD4+/CD8– T cells are present among the neoplastic cells of AITL.70, 71, 72, 73 The reduced or absent expression of pan-T-cell antigen(s) (most commonly sCD3, CD4 and CD7) on neoplastic cells is frequently observed.74 Co-expression of CD10 is observed in a variable proportion of neoplastic cells. Partial CD30 expression is common.75, 76 AITL has been reported to derive from the unique follicular helper T cells subset located in the germinal center. Viruses have been identified as playing a role in the follicular helper T cell transformation. Accordingly, AITL neoplastic cells may express several follicular helper T cell markers, including CXCL13 (a cytoplasmic chemokine), PD1 (a member of the CD28 costimulatory receptor family, resulting in negative regulation of T-cell activity), ICOS (a CD28 homologue with costimulatory function in T-cell activation and expansion) and Bcl-6.77, 78, 79, 80, 81, 82 However, follicular helper T cell markers are not exclusive to AITL. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma cells may also express the Bcl-6+ PD1+ CXCL13+ immunophenotype.83
Extranodal NK/T-cell lymphoma, nasal type
Extranodal NK/T-cell lymphoma, nasal type (ENKTL) is rare in Western countries but relatively common in East Asia (especially China) and Latin America.84 These tumors predominantly occur in extranodal sites, including the nasal or paranasal areas (nasal ENKTL), and less frequently in extranasal sites such as the skin, soft tissue, gastrointestinal tract, testis and brain (extranasal ENKTL).85, 86, 87 Progressive necrotic ulceration and granulation are common in the nasal cavity and midline facial tissues, and nasal obstruction or nasal bleeding due to a mass lesion is the most common symptom at diagnosis.88 Moreover, systemic symptoms such as prolonged fever and weight loss are commonly observed. However, once the tumor develops beyond the original site, the disease rapidly progresses; lymphoma-associated hemophagocytic syndrome is normally noted in such cases and leads rapidly to a fatal outcome.89
A diagnosis of this lymphoma should be considered, particularly if patients present with likely symptoms in a high-prevalence area.90 ENKTL is morphologically heterogeneous, with a cytological spectrum ranging from monomorphic small/medium-sized to large-cell lymphomas, angioinvasive or angiodestructive lymphoid infiltrate, and frequent evidence of necrosis and apoptosis with a heavy admixture of inflammatory cells.91
The detection of CD56 and EBER-1 in tumor cells is important for a diagnosis of ENKTL because these molecules are rarely observed in inflammatory cells within the lesions.87 An invariable association has been demonstrated between ENKTL and EBV.88 ENKTL nasal type was reported to associate strongly with EBV in Asian populations, but the strength of this association in Caucasian populations is less clear.88 However, more than 90% of reported cases were positive for EBNA-1 and EBER-1 (Figure 4). The majority of tumor cells also expressed LMP2. High levels of serum EBV-DNA copy numbers were reported to associate with disease progression and prognosis. In addition to EBV latency proteins, lymphoma cells express other NK-cell markers, including CD2, cytoplasmic CD3 and CD7.92, 93 ENKL is also positive for cytotoxic molecules such as TIA-1, granzyme B and perforin.94

Treatment strategies for inactivating EBV infection or EBV-associated oncogenic pathways. (1) Conventional chemotherapeutic agents or radiation target DNA. (2) Different strategies to disrupt microRNAs can be offered. (3) EBV-specific cytotoxic T cells are used as an immunoregulatory therapeutic approach. (4) Agents can target the lytic cycle of EBV. Agents targeting the (5) NF-kB pathway, (6), PI3K/Akt pathway, (7) PKC pathway, (8) MAPK pathway, (9) Lyn or (10) Sky can be tried. The monoclonal antibody SGNE-35 targets (11) CD30.
The differentiation of NK cells from T-cell lymphoma can be evaluated by the surface expression of CD3, CD5 and TCR on lymphoma cells, as ENKL is negative for these markers.95
Aggressive NK-cell leukemia
Aggressive NK-cell leukemia (ANKL) is a rare malignant disorder of mature NK cells characterized by an aggressive clinical course and poor outcome. Commonly involved sites are the peripheral blood, bone marrow, liver and spleen, but involvement can also occur in other organs.96, 97
The majority of ANKL cases are EBV-positive, and only 10% of reported cases were EBV-negative. The immunophenotypic findings are almost identical to those of ENKL, nasal type, which has a leukemic phase.98 However, CD16 expression is characteristic for ANKL, compared with ENKL.99 Furthermore, surface CD3 negativity, as determined by flow cytometric or immunophenotypic analysis, and germline TCR gene configurations, as determined in TCR rearrangement studies, can differentiate T-cell-type large granular lymphocytic leukemia or leukemic infiltrations of other T-cell lymphomas from ANKL.96 Recurrent chromosomal abnormalities such as a gain of 1q and losses of 7p15.1–p22.3 and 17p13.1 are characteristic in ANKL.100
T-cell LPDs in children
The current World Health Organization (WHO) classification includes two major types of EBV-positive T-cell LPDs in children: systemic EBV-positive T-cell LPD of childhood, and hydroa vacciniforme-like lymphoma with a variable clinical course. Systemic EBV-positive T-cell LPD of childhood is extremely rare and has an aggressive clinical course. The majority of these cases occur with an acute EBV infection. The typical phenotype is CD2+, CD3+, CD8+ and TIA-1+. CD56 is usually not expressed. In this disease, cytological atypia of neoplastic cells is minimal; double staining for EBER1 and CD3 or CD8 is useful for diagnosis.101, 102, 103
Infantile fulminant EBV-positive T-LPD, which was designated as systemic T-cell LPD of childhood in the 2008 WHO classification, is characterized by rapid deterioration in infants within a few days or weeks after a primary acute EBV infection and is accompanied by hemophagocytic syndrome. Such patients may have a high fever, skin rash and jaundice. Patients also present with pulmonary infiltrate, hepatosplenomegaly, pancytopenia, coagulopathy and abnormal liver function.104, 105 The clinical course is characterized by rapid deterioration, with the main causes of death being coagulopathy, multiple organ failure and opportunistic infection. Bone marrow studies have revealed infiltration by atypical T lymphocytes and rare B immunoblasts, as well as mature histiocytes with hemophagocytosis. Of special interest is that in this disease, the presence of EBV has been detected exclusively in T lymphoid cells, which were often genetically confirmed to have undergone clonal proliferation.105, 106, 107
Hydroa vacciniforme-like lymphoma occurs mainly in Central and South America and Asia.108 There is strong evidence for a pathogenic relationship between hydroa vacciniforme-like lymphoma and ultraviolet light exposure. Patients are often found to have necrotic vesiculopapules on exposed areas, and the disease has a chronic clinical course with worsening cutaneous symptoms and eventual systemic dissemination.109 Histologically, this disease is usually characterized by polymorphic lymphocyte infiltration of the dermis. The cells are CD8+ and exhibit monoclonal TCR gene rearrangement. There is also a strong association with EBV infection.109, 110
Chronic active EBV infection
Chronic active EBV infection (CAEBV) was originally related to chronic or persistent EBV infection. On the basis of the EBV-induced clonal expansion of different lymphocytes, the origin of CAEBV is classified as B, T or NK cell. CAEBV, B-cell type is very rare in comparison with the T-cell type.111
Patients with CAEBV present with fever, splenomegaly, lymphadenopathy, hepatic dysfunction and pancytopenia,111 and have markedly elevated peripheral blood levels of EBV DNA. This disease is rare with variable clinical severity and often causes high morbidity and mortality. Histologically, the lymph node pathology is variable and often similar to that of a polymorphic PTLD with paracortical expansion and numerous immunoblasts. Cells with plasmacytoid differentiation, plasma cells and occasional Reed–Sternberg-like cells are often observed. Variable numbers of the infiltrating cells are positive for EBERs.111, 112
Hypersensitivity to mosquito bites
Hypersensitivity to mosquito bites is a rare disease characterized by severe local skin reactions and general symptoms such as high fever, liver dysfunction, high IgE levels and regional lymphadenopathy after mosquito bites.113, 114 NK-cell lymphocytosis is frequently observed in the peripheral blood. The mean age of onset is 6.7 years, with no gender predominance. hypersensitivity to mosquito bites is usually associated with chronic EBV infection as well as NK-cell leukemia/lymphoma.114 Mosquito bite-stimulated CD4+ T cells might associate with the development of hypersensitivity to mosquito bites and NK-cell oncogenesis by inducing EBV reactivation and EBV-oncogene expression, respectively. Hypersensitivity to mosquito bites patients without systemic symptoms may eventually develop CAEBV.115 Spongiotic epidermis and a polymorphous cellular infiltrate with angiocentricity throughout the dermis may be observed with this disease.
EBV-positive HLH
EBV-positive HLH includes a broad spectrum of diseases ranging from EBV-associated reactive, polyclonal LPD to monoclonal diseases. The most typical clinical presentations of HLH are fever, hepatosplenomegaly and cytopenia. According to the HLH-2004 guidelines, patients must fulfill five of the following eight criteria: (i) fever; (ii) splenomegaly; (iii) cytopenia affecting at least two of three lineages in the peripheral blood; (iv) hypertriglyceridemia and/or hypofibrinogenemia; (v) hemophagocytosis in the bone marrow, spleen or lymph nodes; (vi) low or absent NK-cell activity; (vii) hyperferritinemia and (viii) high levels of sIL-2R.116 TCR gamma and immunoglobulin heavy chain gene rearrangement have no clinical significance in patients with HLH. However, a high EBV-DNA load may be a risk factor for a poor outcome.117
T-cell posttransplant lymphoproliferative disorder (T-cell PTLD)
PTLDs are a heterogeneous group of LPDs that arise after solid organ and other transplantations. According to the 2008 WHO classification, PTLD can be subclassified as early lesions, polymorphic PTLD, monomorphic B- or T-cell PTLD and Hodgkin’s-type PTLD. T-PTLDs manifest as a variety of aberrant T-cell proliferation disorders, and patients exhibit a uniformly poor prognosis.
PTLDs are classically of B-cell origin; T-cell PTLD following hematopoietic stem cell transplantation is exceedingly rare. However, a recent study reported that 7–15% of PTLD were of T-cell and NK-cell origin.118 More than 90% of all PTLD cases have been linked to EBV. However, the rate of EBV association is much higher among B-cell PTLDs (80% in late onset lesions, 100% in early onset lesions) than among non B-cell tumors (37%). Therefore, some experts have suggested that T lymphocytes do not express the EBV receptor CD21. However, one-third of T-PTLD cases have been reported to comprise aberrant T cells that are positive for CD21 and EBV.119 Other viruses such as CMV, HTLV and HHV-6 were also detectable and occurred as co-infections with EBV in some patients. EBV viral load monitoring is a routinely used and powerful tool for EBV detection and estimates the risk for PTLD development. However, virus seropositivity might be associated with immunosuppression rather than the initiating cause in all cases of T-PTLD.
The clinical presentation is variable and depends on the underlying pathologic condition, type, interval since transplant and time of duration since transplant. Among transplanted organs, the kidney is most frequently affected by both monomorphic T-PTLD and B-PTLD.20, 119, 120
Treatment of EBV-positive T/NK-cell LPD
In cases of localized T/NK-cell LPD, cytotoxic chemotherapy and/or local radiotherapy are frequently used therapeutic options. However, EBV-associated T/NK-cell LPD frequently expresses a P-glycoprotein, leading to chemotherapy resistance in the majority of cases. L-asparaginase and high-dose cytarabine (Ara-C) are reportedly effective in patients with resistant or relapsed disease.121
Although some patients may transiently respond to immunosuppressive and cytotoxic chemotherapies, others especially patients with advanced disease, may be unresponsive. Therefore, autologous or allogeneic hematopoietic stem cell transplantation seems a feasible and promising method for curing relapsed or advanced-stage EBV-associated T/NK-cell LPD. As EBV-specific T-cell-based and monoclonal antibodies are being used to treat posttransplantation B-cell LPD, in which target EBV antigens such as the EBNAs and LMPs and the B-cell antigen (CD20) are expressed, a similar approach appears possible for T/NK-cell LPD.121
For several decades, the reduction or cessation of immune suppression has been used as a first-line treatment for EBV and PTLD. However, adoptive cellular immunotherapy has recently been very successful in some circumstances and offers the potential of overcoming cellular resistance to chemotherapy. Antiviral drugs such as acyclovir and ganciclovir can be used as an adjunctive therapy.122, 123 However, the current treatment for most EBV-associated T/NK-cell LPD is unsatisfactory, and therapies involving novel mechanisms that target important signaling pathways are critically required.
Conclusions
EBV-associated T/NK-cell LPDs comprise a heterogeneous group of diseases that occur consequent to defects in the cellular immune system. The underlying pathogenesis is complex and includes genetic abnormalities in T or NK cells. The precise role played by EBV in lymphomagenesis remains unclear. LPDs derived from T cells and NK cells often exhibit overlapping clinical symptoms as well as histologic and immunophenotypic features because both types of lymphoid cells arise from a common developmental precursor. A better understanding of the pathogenesis and its relationship with clinical manifestations is necessary for developing strategies to control the ectopic EBV infections that underlie these unique syndromes.
References
Vallejo AN, Davila E, Weyand CM, Goronzy JJ . Biology of T lymphocytes. Rheum Dis Clin North Am 2004; 30: 135–157.
Campbell KS, Hasegawa J . Natural killer cell biology: an update and future directions. J Allergy Clin Immunol 2013; 132: 536–544.
Morice WG . The immunophenotypic attributes of NK cells and NK-cell lineage lymphoproliferative disorders. Am J Clin Pathol 2007; 127: 881–886.
Lanier LL, Spits H, Phillips JH . The developmental relationship between NK cells and T cells. Immunol Today 1992; 13: 392–395.
Delecluse HJ, Feederle R, O'Sullivan B, Taniere P . Epstein Barr virus-associated tumours: an update for the attention of the working pathologist. J Clin Pathol 2007; 60: 1358–1364.
Rickinson AB . Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol 2014; 26: 99–115.
Parvaneh N, Filipovich AH, Borkhardt A . Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol 2013; 162: 573–586.
Chang CM, Yu KJ, Mbulaiteye SM, Hildesheim A, Bhatia K . The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus Res 2009; 143: 209–221.
Rickinson AB, Kieff E . Epstein–Barr virus. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (eds) Fields Virology, 5th edition. Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007, pp 2655–2700.
Merlo A, Turrini R, Dolcetti R, Martorelli D, Muraro E, Comoli P et al. The interplay between Epstein-Barr virus and the immune system: a rationale for adoptive cell therapy of EBV-related disorders. Haematologica 2010; 95: 1769–1777.
Toczyski DP, Matera AG, Ward DC, Steitz JA . The Epstein-Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc Natl Acad Sci USA 1994; 91: 3463–3467.
Cesarman E . Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 2014; 9: 349–372.
Petrara MR, Freguja R, Gianesin K, Zanchetta M, De Rossi A . Epstein-Barr virus-driven lymphomagenesis in the context of human immunodeficiency virus type 1 infection. Front Microbiol 2013; 4: 311.
Roschewski M, Wilson WH . EBV-associated lymphomas in adults. Best Pract Res Clin Haematol 2012; 25: 75–89.
Young LS, Murray PG . Epstein-Barr virus and oncogenesis: from latent genes to tumours. Oncogene 2003; 22: 5108–5121.
Saha A, Robertson ES . Epstein-Barr virus-associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res 2011; 17: 3056–3063.
Chen J . Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J Virol 2012; 1: 154–161.
Lee JW, Liu PF, Hsu LP, Chen PR, Chang CH, Shih WL . EBV LMP-1 negatively regulates expression and pro-apoptotic activity of Par-4 in nasopharyngeal carcinoma cells. Cancer Lett 2009; 279: 193–201.
Vrzalikova K, Vockerodt M, Leonard S, Bell A, Wei W, Schrader A et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011; 117: 5907–5917.
Nourse JP, Jones K, Gandhi MK . Epstein-Barr virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transplant 2011; 11: 888–895.
Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W . Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog 2010; 6: e1001063.
Hong GK, Kumar P, Wang L, Damania B, Gulley ML, Delecluse HJ et al. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J Virol 2005; 79: 13984–13992.
Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 2011; 85: 165–177.
Burns DM, Crawford DH . Epstein-Barr virus-specific cytotoxic T-lymphocytes for adoptive immunotherapy of post-transplant lymphoproliferative disease. Blood Rev 2004; 18: 193–209.
Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med 1994; 330: 1185–1191.
Li J, Qian CN, Zeng YX . Regulatory T cells and EBV associated malignancies. Int Immunopharmacol 2009; 9: 590–592.
Menasche G, Feldmann J, Fischer A, de Saint Basile G . Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev 2005; 203: 165–179.
Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med 2011; 3: 581–592.
Grivennikov SI, Greten FR, Karin M . Immunity, inflammation, and cancer. Cell 2010; 140: 883–899.
Balkwill FR, Mantovani A . Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 2012; 22: 33–40.
Pittaluga S . Viral-associated lymphoid proliferations. Semin Diagn Pathol 2013; 30: 130–136.
Terrin L, Dal Col J, Rampazzo E, Zancai P, Pedrotti M, Ammirabile G et al. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J Virol 2008; 82: 10175–10187.
Terrin L, Dolcetti R, Corradini I, Indraccolo S, Dal Col J, Bertorelle R et al. hTERT inhibits the Epstein-Barr virus lytic cycle and promotes the proliferation of primary B lymphocytes: implications for EBV-driven lymphomagenesis. Int J Cancer 2007; 121: 576–587.
Michelow P, Wright C, Pantanowitz L . A review of the cytomorphology of Epstein-Barr virus-associated malignancies. Acta Cytol 2012; 56: 1–14.
Gandhi MK . Epstein-Barr virus-associated lymphomas. Expert Rev Anti Infect Ther 2006; 4: 77–89.
Shah KM, Young LS . Epstein-Barr virus and carcinogenesis: beyond Burkitt's lymphoma. Clin Microbiol Infect 2009; 15: 982–988.
Cardenas-Mondragon MG, Carreon-Talavera R, Camorlinga-Ponce M, Gomez-Delgado A, Torres J, Fuentes-Panana EM . Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PloS One 2013; 8: e62850.
McClain KL, Leach CT, Jenson HB, Joshi VV, Pollock BH, Parmley RT et al. Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 1995; 332: 12–18.
Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG . The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A 2009; 106: 2313–2318.
Gruhne B, Sompallae R, Masucci MG . Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009; 28: 3997–4008.
Harabuchi Y, Takahara M, Kishibe K, Moriai S, Nagato T, Ishii H . Nasal natural killer (NK)/T-cell lymphoma: clinical, histological, virological, and genetic features. Int J Clin Oncol 2009; 14: 181–190.
Hui AB, Lo KW, Teo PM, To KF, Huang DP . Genome wide detection of oncogene amplifications in nasopharyngeal carcinoma by array based comparative genomic hybridization. Int J Oncol 2002; 20: 467–473.
Wong N, Hui AB, Fan B, Lo KW, Pang E, Leung SF et al. Molecular cytogenetic characterization of nasopharyngeal carcinoma cell lines and xenografts by comparative genomic hybridization and spectral karyotyping. Cancer Genet Cytogenet 2003; 140: 124–132.
Cesarman E . Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 2011; 305: 163–174.
Cesarman E, Chadburn A, Liu YF, Migliazza A, Dalla-Favera R, Knowles DM . BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood 1998; 92: 2294–2302.
Wensing B, Farrell PJ . Regulation of cell growth and death by Epstein-Barr virus. Microbes Infect 2000; 2: 77–84.
Capello D, Rossi D, Gaidano G . Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol 2005; 23: 61–67.
Schuster V, Ott G, Seidenspinner S, Kreth HW . Common Epstein-Barr virus (EBV) type-1 variant strains in both malignant and benign EBV-associated disorders. Blood 1996; 87: 1579–1585.
Taguchi A, Miyazaki M, Sakuragi S, Shinohara K, Kamei T, Inoue Y . Gamma/delta T cell lymphoma. Intern Med 2004; 43: 120–125.
Volanakis EJ, Boothby MR, Sherr CJ . Epigenetic regulation of the Ink4a-Arf (Cdkn2a) tumor suppressor locus in the initiation and progression of Notch1-driven T cell acute lymphoblastic leukemia. Exp Hematol 2013; 41: 377–386.
Fasseu M, Aplan PD, Chopin M, Boissel N, Bories JC, Soulier J et al. p16INK4A tumor suppressor gene expression and CD3epsilon deficiency but not pre-TCR deficiency inhibit TAL1-linked T-lineage leukemogenesis. Blood 2007; 110: 2610–2619.
Tsellou E, Troungos C, Moschovi M, Athanasiadou-Piperopoulou F, Polychronopoulou S, Kosmidis H et al. Hypermethylation of CpG islands in the promoter region of the p15INK4B gene in childhood acute leukaemia. Eur J Cancer 2005; 41: 584–589.
Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K . Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci USA 2011; 108: 1919–1924.
Skalska L, White RE, Parker GA, Sinclair AJ, Paschos K, Allday MJ . Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog 2013; 9: e1003187.
Campo E, Gaulard P, Zucca E, Jaffe ES, Harris NL, Diebold J et al. Report of the European Task Force on Lymphomas: workshop on peripheral T-cell lymphomas. Ann Oncol 1998; 9: 835–843.
Falini B, Pileri S, De Solas I, Martelli MF, Mason DY, Delsol G et al. Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood 1990; 75: 434–444.
Vose J, Armitage J, Weisenburger D . International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26: 4124–4130.
Gallamini A, Stelitano C, Calvi R, Bellei M, Mattei D, Vitolo U et al. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood 2004; 103: 2474–2479.
Quintanilla-Martinez L, Fend F, Moguel LR, Spilove L, Beaty MW, Kingma DW et al. Peripheral T-cell lymphoma with Reed-Sternberg-like cells of B-cell phenotype and genotype associated with Epstein-Barr virus infection. Am J Surg Pathol 1999; 23: 1233–1240.
Mendonca MC, Doi SQ, Glerum S, Sellitti DF . Increase of C-type natriuretic peptide expression by serum and platelet-derived growth factor-BB in human aortic smooth muscle cells is dependent on protein kinase C activation. Endocrinology 2006; 147: 4169–4178.
Went P, Agostinelli C, Gallamini A, Piccaluga PP, Ascani S, Sabattini E et al. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J Clin Oncol 2006; 24: 2472–2479.
Attygalle AD, Kyriakou C, Dupuis J, Grogg KL, Diss TC, Wotherspoon AC et al. Histologic evolution of angioimmunoblastic T-cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression. Am J Surg Pathol 2007; 31: 1077–1088.
Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ . Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am J Surg Pathol 2006; 30: 802–810.
Grogg KL, Attygalle AD, Macon WR, Remstein ED, Kurtin PJ, Dogan A . Expression of CXCL13, a chemokine highly upregulated in germinal center T-helper cells, distinguishes angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified. Mod Pathol 2006; 19: 1101–1107.
Gisselbrecht C, Lepage E, Molina T, Quesnel B, Fillet G, Lederlin P et al. Shortened first-line high-dose chemotherapy for patients with poor-prognosis aggressive lymphoma. J Clin Oncol 2002; 20: 2472–2479.
Mounier N, Gisselbrecht C, Briere J, Haioun C, Feugier P, Offner F et al. Prognostic factors in patients with aggressive non-Hodgkin's lymphoma treated by front-line autotransplantation after complete remission: a cohort study by the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2004; 22: 2826–2834.
Rudiger T, Weisenburger DD, Anderson JR, Armitage JO, Diebold J, MacLennan KA et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin's Lymphoma Classification Project. Ann Oncol 2002; 13: 140–149.
de Leval L, Gisselbrecht C, Gaulard P . Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol 2010; 148: 673–689.
Cho YU, Chi HS, Park CJ, Jang S, Seo EJ, Huh J . Distinct features of angioimmunoblastic T-cell lymphoma with bone marrow involvement. Am J Clin Pathol 2009; 131: 640–646.
Attygalle A, Al-Jehani R, Diss TC, Munson P, Liu H, Du MQ et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood 2002; 99: 627–633.
Patsouris E, Noel H, Lennert K . Angioimmunoblastic lymphadenopathy—type of T-cell lymphoma with a high content of epithelioid cells. Histopathology and comparison with lymphoepithelioid cell lymphoma. Am J Surg Pathol 1989; 13: 262–275.
Willenbrock K, Renne C, Gaulard P, Hansmann ML . In angioimmunoblastic T-cell lymphoma, neoplastic T cells may be a minor cell population. A molecular single-cell and immunohistochemical study. Virchows Arch 2005; 446: 15–20.
Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K . Peripheral T-cell lymphoma. Blood 2011; 117: 6756–6767.
Willenbrock K, Roers A, Seidl C, Wacker HH, Kuppers R, Hansmann ML . Analysis of T-cell subpopulations in T-cell non-Hodgkin's lymphoma of angioimmunoblastic lymphadenopathy with dysproteinemia type by single target gene amplification of T cell receptor- beta gene rearrangements. Am J Pathol 2001; 158: 1851–1857.
Karube K, Aoki R, Nomura Y, Yamamoto K, Shimizu K, Yoshida S et al. Usefulness of flow cytometry for differential diagnosis of precursor and peripheral T-cell and NK-cell lymphomas: analysis of 490 cases. Pathol Int 2008; 58: 89–97.
Attygalle AD, Chuang SS, Diss TC, Du MQ, Isaacson PG, Dogan A . Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype and molecular genetics. Histopathology 2007; 50: 498–508.
Rodriguez-Justo M, Attygalle AD, Munson P, Roncador G, Marafioti T, Piris MA . Angioimmunoblastic T-cell lymphoma with hyperplastic germinal centres: a neoplasia with origin in the outer zone of the germinal centre? Clinicopathological and immunohistochemical study of 10 cases with follicular T-cell markers. Mod Pathol 2009; 22: 753–761.
Grogg KL, Attygalle AD, Macon WR, Remstein ED, Kurtin PJ, Dogan A . Angioimmunoblastic T-cell lymphoma: a neoplasm of germinal-center T-helper cells? Blood 2005; 106: 1501–1502.
Roncador G, Garcia Verdes-Montenegro JF, Tedoldi S, Paterson JC, Klapper W, Ballabio E et al. Expression of two markers of germinal center T cells (SAP and PD-1) in angioimmunoblastic T-cell lymphoma. Haematologica 2007; 92: 1059–1066.
Krenacs L, Schaerli P, Kis G, Bagdi E . Phenotype of neoplastic cells in angioimmunoblastic T-cell lymphoma is consistent with activated follicular B helper T cells. Blood 2006; 108: 1110–1111.
Marafioti T, Paterson JC, Ballabio E, Chott A, Natkunam Y, Rodriguez-Justo M et al. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica 2010; 95: 432–439.
Yuan CM, Vergilio JA, Zhao XF, Smith TK, Harris NL, Bagg A . CD10 and BCL6 expression in the diagnosis of angioimmunoblastic T-cell lymphoma: utility of detecting CD10+ T cells by flow cytometry. Hum Pathol 2005; 36: 784–791.
Rodriguez Pinilla SM, Roncador G, Rodriguez-Peralto JL, Mollejo M, Garcia JF, Montes-Moreno S et al. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma expresses follicular T-cell markers. Am J Surg Pathol 2009; 33: 81–90.
Guo HQ, Huang GL, Guo CC, Pu XX, Lin TY . Diagnostic and prognostic value of circulating miR-221 for extranodal natural killer/T-cell lymphoma. Dis Markers 2010; 29: 251–258.
Oshimi K . Progress in understanding and managing natural killer-cell malignancies. Br J Haematol 2007; 139: 532–544.
Harabuchi Y, Imai S, Wakashima J, Hirao M, Kataura A, Osato T et al. Nasal T-cell lymphoma causally associated with Epstein-Barr virus: clinicopathologic, phenotypic, and genotypic studies. Cancer 1996; 77: 2137–2149.
Hirakawa S, Kuyama M, Takahashi S, Yamasaki O, Kanzaki H, Teshima T et al. Nasal and nasal-type natural killer/T-cell lymphoma. J Am Acad Dermatol 1999; 40: 268–272.
Harabuchi Y, Yamanaka N, Kataura A, Imai S, Kinoshita T, Mizuno F et al. Epstein-Barr virus in nasal T-cell lymphomas in patients with lethal midline granuloma. Lancet 1990; 335: 128–130.
Takahashi N, Miura I, Chubachi A, Miura AB, Nakamura S . A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol 2001; 74: 303–308.
Liang R . Diagnosis and management of primary nasal lymphoma of T-cell or NK-cell origin. Clin Lymphoma 2000; 1: 33–37; discussion 38.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–1392.
Mori N, Yatabe Y, Oka K, Kinoshita T, Kobayashi T, Ono T et al. Expression of perforin in nasal lymphoma. Additional evidence of its natural killer cell derivation. Am J Pathol 1996; 149: 699–705.
Suzumiya J, Takeshita M, Kimura N, Kikuchi M, Uchida T, Hisano S et al. Expression of adult and fetal natural killer cell markers in sinonasal lymphomas. Blood 1994; 83: 2255–2260.
Li CC, Tien HF, Tang JL, Yao M, Chen YC, Su IJ et al. Treatment outcome and pattern of failure in 77 patients with sinonasal natural killer/T-cell or T-cell lymphoma. Cancer 2004; 100: 366–375.
Emile JF, Boulland ML, Haioun C, Kanavaros P, Petrella T, Delfau-Larue MH et al. CD5-CD56+ T-cell receptor silent peripheral T-cell lymphomas are natural killer cell lymphomas. Blood 1996; 87: 1466–1473.
Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S et al. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 2004; 18: 763–770.
Ishida F, Ko YH, Kim WS, Suzumiya J, Isobe Y, Oshimi K et al. Aggressive natural killer cell leukemia: therapeutic potential of L-asparaginase and allogeneic hematopoietic stem cell transplantation. Cancer Sci 2012; 103: 1079–1083.
Ryder J, Wang X, Bao L, Gross SA, Hua F, Irons RD . Aggressive natural killer cell leukemia: report of a Chinese series and review of the literature. Int J Hematol 2007; 85: 18–25.
Caligiuri MA . Human natural killer cells. Blood 2008; 112: 461–469.
Lima M . Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis 2013; 8: 95.
Kimura H, Ito Y, Kawabe S, Gotoh K, Takahashi Y, Kojima S et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 2012; 119: 673–686.
Kasahara Y, Yachie A, Takei K, Kanegane C, Okada K, Ohta K et al. Differential cellular targets of Epstein-Barr virus (EBV) infection between acute EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Blood 2001; 98: 1882–1888.
Fox CP, Shannon-Lowe C, Gothard P, Kishore B, Neilson J, O'Connor N et al. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults characterized by high viral genome load within circulating natural killer cells. Clin Infect Dis 2010; 51: 66–69.
Su IJ, Lin DT, Hsieh HC, Lee SH, Chen J, Chen RL et al. Fatal primary Epstein-Barr virus infection masquerading as histiocytic medullary reticulosis in young children in Taiwan. Hematol Pathol 1990; 4: 189–195.
Chen RL, Su IJ, Lin KH, Lee SH, Lin DT, Chuu WM et al. Fulminant childhood hemophagocytic syndrome mimicking histiocytic medullary reticulosis. An atypical form of Epstein-Barr virus infection. Am J Clin Pathol 1991; 96: 171–176.
Suzuki K, Ohshima K, Karube K, Suzumiya J, Ohga S, Ishihara S et al. Clinicopathological states of Epstein-Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection) of children and young adults. Int J Oncol 2004; 24: 1165–1174.
Su IJ, Chen RL, Lin DT, Lin KS, Chen CC . Epstein-Barr virus (EBV) infects T lymphocytes in childhood EBV-associated hemophagocytic syndrome in Taiwan. Am J Pathol 1994; 144: 1219–1225.
Quintanilla-Martinez L, Ridaura C, Nagl F, Saez-de-Ocariz M, Duran-McKinster C, Ruiz-Maldonado R et al. Hydroa vacciniforme-like lymphoma: a chronic EBV+ lymphoproliferative disorder with risk to develop a systemic lymphoma. Blood 2013; 122: 3101–3110.
Iwatsuki K, Xu Z, Takata M, Iguchi M, Ohtsuka M, Akiba H et al. The association of latent Epstein-Barr virus infection with hydroa vacciniforme. Br J Dermatol 1999; 140: 715–721.
Cho KH, Lee SH, Kim CW, Jeon YK, Kwon IH, Cho YJ et al. Epstein-Barr virus-associated lymphoproliferative lesions presenting as a hydroa vacciniforme-like eruption: an analysis of six cases. Br J Dermatol 2004; 151: 372–380.
Kimura H, Hoshino Y, Kanegane H, Tsuge I, Okamura T, Kawa K et al. Clinical and virologic characteristics of chronic active Epstein-Barr virus infection. Blood 2001; 98: 280–286.
Cohen JI, Kimura H, Nakamura S, Ko YH, Jaffe ES . Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol 2009; 20: 1472–1482.
Ishihara S, Yabuta R, Tokura Y, Ohshima K, Tagawa S . Hypersensitivity to mosquito bites is not an allergic disease, but an Epstein-Barr virus-associated lymphoproliferative disease. Int J Hematol 2000; 72: 223–228.
Tokura Y, Ishihara S, Tagawa S, Seo N, Ohshima K, Takigawa M . Hypersensitivity to mosquito bites as the primary clinical manifestation of a juvenile type of Epstein-Barr virus-associated natural killer cell leukemia/lymphoma. J Am Acad Dermatol 2001; 45: 569–578.
Kawa K, Okamura T, Yagi K, Takeuchi M, Nakayama M, Inoue M . Mosquito allergy and Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disease. Blood 2001; 98: 3173–3174.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48: 124–131.
Ahn JS, Rew SY, Shin MG, Kim HR, Yang DH, Cho D et al. Clinical significance of clonality and Epstein-Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2010; 85: 719–722.
Lau LG, Tan LK, Salto-Tellez M, Koay ES, Liu TC . T-cell post-transplant lymphoproliferative disorder after hematopoietic stem cell transplantation: another case and a review of the literature. Bone Marrow Transplant 2004; 34: 821–822.
Tiede C, Maecker-Kolhoff B, Klein C, Kreipe H, Hussein K . Risk factors and prognosis in T-cell posttransplantation lymphoproliferative diseases: reevaluation of 163 cases. Transplantation 2013; 95: 479–488.
Green M, Michaels MG . Epstein-Barr virus infection and posttransplant lymphoproliferative disorder. Am J Transplant. 2013; 13 (Suppl 3): 41–54; quiz 54.
Kawa K . Diagnosis and treatment of Epstein-Barr virus-associated natural killer cell lymphoproliferative disease. Int J Hematol 2003; 78: 24–31.
Bollard CM, Rooney CM, Heslop HE . T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol 2012; 9: 510–519.
Hatton O, Martinez OM, Esquivel CO . Emerging therapeutic strategies for Epstein-Barr virus+ post-transplant lymphoproliferative disorder. Pediatr Transplant 2012; 16: 220–229.
Acknowledgements
QC is the recipient of funding from the National Natural Science Foundation of China (NO.81372883, NO.81001052), Science and Technology Planning Project of Guangdong Province, China (2011B031800222), Young Talents Project of Sun Yat-sen University Cancer Center, Young Talents Project of Sun Yat-sen University and the Natural Science Foundation of Guangdong Province, China (8151008901000043). KHY is supported by The University of Texas MD Anderson Cancer Center Institutional Research and Development Fund, Institutional Research Grant Award, an MD Anderson Cancer Center Lymphoma SPORE Research Development Program Award, an MD Anderson Cancer Center Myeloma SPORE Research Development Program Award, Gundersen Lutheran Medical Foundation Award, MD Anderson Cancer Center Collaborative Funds with Roche Molecular System, Gilead Science, and HTG Molecular Diagnostics. This study was supported by the National Cancer Institute/National Institutes of Health (R01CA138688, 1RC1CA146299, P50CA136411 and P50CA142509).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Cai, Q., Chen, K. & Young, K. Epstein–Barr virus-positive T/NK-cell lymphoproliferative disorders. Exp Mol Med 47, e133 (2015). https://doi.org/10.1038/emm.2014.105
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.105
This article is cited by
-
Tumor resident, TRA anti-viral CDR3 chemical sequence motifs are associated with a better breast cancer outcome
Genes & Immunity (2023)
-
Familial hemophagocytic lymphohistiocytosis with Epstein–Barr virus infection and progression to aggressive NK-cell leukemia: a case report and review of the literature
Journal of Hematopathology (2022)
-
Multiple small tumor formation on both surfaces of the aortic valve cusps in Epstein–Barr virus-associated T/NK-cell lymphoproliferative disease: a case report
General Thoracic and Cardiovascular Surgery (2021)
-
Pathogenesis and biomarkers of natural killer T cell lymphoma (NKTL)
Journal of Hematology & Oncology (2019)
-
Molecular pathogenic pathways in extranodal NK/T cell lymphoma
Journal of Hematology & Oncology (2019)
