
Abstract
We previously reported that the p53 tumor suppressor protein plays an essential role in the induction of tetraploid G1 arrest in response to perturbation of the actin cytoskeleton, termed actin damage. In this study, we investigated the role of p53, ataxia telangiectasia mutated protein (ATM), and catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) in tetraploid G1 arrest induced by actin damage. Treatment with actin-damaging agents including pectenotoxin-2 (PTX-2) increases phosphorylation of Ser-15 and Ser-37 residues of p53, but not Ser-20 residue. Knockdown of ATM and DNA-PKcs do not affect p53 phosphorylation induced by actin damage. However, while ATM knockdown does not affect tetraploid G1 arrest, knockdown of DNA-PKcs not only perturbs tetraploid G1 arrest, but also results in formation of polyploidy and induction of apoptosis. These results indicate that DNA-PKcs is essential for the maintenance of actin damage induced-tetraploid G1 arrest in a p53-independent manner. Furthermore, actin damage-induced p53 expression is not observed in cells synchronized at G1/S of the cell cycle, implying that p53 induction is due to actin damage-induced tetraploidy rather than perturbation of actin cytoskeleton. Therefore, these results suggest that p53 and DNA-PKcs independently function for tetraploid G1 arrest and preventing polyploidy formation.
Similar content being viewed by others
Introduction
Various cell cycle checkpoints ensure coordinated progression of the cell cycle (Hartwell and Weinert, 1989) and checkpoint defects result in gene mutations, chromosome damage, and aneuploidy, all of which can contribute to tumorigenesis (Paulovich et al., 1997). A molecular understanding of various cellular checkpoints should therefore provide a basis for the development of new therapeutic approaches for many cancers with abnormalities in checkpoint functions (Stewart and Pietenpol, 1999). The G1 checkpoint permits repair prior to replication, whereas the G2 checkpoint allows repair of the genome prior to its mitotic segregation. The p53 tumor suppressor gene, which is suppressed by mutation in approximately one-half of human tumors (Hollstein et al., 1991), has been shown to be integral to both the G1 (Kuerbitz et al., 1992) and G2 (Bunz et al., 1998) DNA damage checkpoint machinery. In addition to DNA damage, spindle damage induces a transient arrest at the metaphase-anaphase transition point (Rudner and Murray, 1996; Amon, 1999), but eventually escape from this block and exit mitosis without proper segregation of sister chromatids and cytokinesis (Jordan et al., 1991, 1996; Torres and Horwitz, 1998). Therefore, spindle-damaged cells arrest at a G1-like state with an intact nucleus containing 4N DNA, but without ever completing mitosis (Minn et al., 1996; Lanni and Jacks, 1998). This is termed a tetraploid G1 state.
Actin depolymerizing agents such as pectenotoxin-2 (PTX-2) and dihydrocytochalasin B (DCB), which inhibit cytokinesis by depolymerizing actin filaments, also lead to tetraploid G1 arrest (Andreassen et al., 2001; Chae et al., 2005). Since actin-damaging agents do not affect spindle function and chromatid segregation, but only inhibit cytokinesis, the tetraploid G1 checkpoint seems to be a general checkpoint control acting in G1 to recognize tetraploid cells and induce their arrest and thereby prevent the generation of aneuploidy (Margolis et al., 2003; Margolis, 2005).
We previously reported that loss of p53 sensitizes tumor cells to actin damage induced by treatment with actin-depolymerizing or knotting agents (Chae et al., 2005). Upon actin damage, Bim expression was induced in tumor cells lacking functional p53 followed by conformational changes of Bax protein (Chae et al., 2005). Furthermore, induction of Bimmediated apoptosis by actin damage in p53-deficient cells results from constitutive cdk2 activation and its associated genomic instability (Chae et al., 2008). In this study, we demonstrate that actin damage, like DNA damage, induces phosphorylation of Ser-15 and Ser-37 residues of p53 protein, but the upstream signaling pathways leading to the phosphorylation was different in between DNA and actin damage responses. However, although DNA-PKcs is not responsible for phosphorylation of p53 protein, it was shown to be essential for the maintenance of tetraploid G1 arrest following actin damage and cytokinesis failure.
Results and Discussion
We previously reported that actin damage induces tetraploid G1-arrest by activating the p53 tumor suppressor protein, which leads to inactivation of Cdk2 (Chae et al., 2005, 2008). In this study, we first addressed the molecular mechanism underlying p53 induction by actin damage. Since it has been clearly shown that p53 accumulation is largely dependent on the phosphorylation state of the amino-terminal region of p53 protein (Giaccia and Kastan, 1998; Ashcroft et al., 1999; Oren, 1999), we examined major phosphorylation sites of the N-terminal region of p53. While a DNA damaging agent, doxorubicin, induced phosphorylation of serine residues located at the amino terminal, namely Ser-15, -20, and -37, all of the actin inhibitors tested here induced phosphorylation of Ser-15 and -37, but left Ser-20 unphosphorylated (Figures 1A and 1B).

Phosphorylation of p53 upon actin damage. (A) HCT116 colon carcinoma cells were treated with PTX-2 (100 ng/ml) for 72 h or doxorubicin (150 nM) for 24 h. (B) HCT116 cells were treated with actin-damaging agents, such as PTX-2 (100 ng/ml), cytochalasin D (10 µM), and psychosine (50 µM) were for 72 h. Cell extracts were subjected to Western blot analysis with the indicated antibodies.
It was further examined the effects of caffeine and wortmannin, two PI3 kinase inhibitors known to inhibit p53 phosphorylation upon DNA damage (Ui et al., 1995; Kaufmann et al., 2003), on the phosphorylation of Ser-15 and -37 of p53. Both inhibitors blocked DNA damage-induced phosphorylation of all three serine residues in p53 protein (Figure 2A). However, we found that the PI3 kinase inhibitors function differentially in the phosphorylation of p53 induced by actin damage. While caffeine did not inhibit PTX-2-induced phosphorylation of Ser-15 and Ser-37, wortmannin blocked their phosphorylation (Figure 2B). PD98059 and SB212090, which inhibit ERK1 and p38, respectively, did not affect phosphorylation of Ser-15 and Ser-37 (Figure 2C). Thus, these results suggest that a caffeine-insensitive, but a wortmannin-sensitive kinase is responsible for actin damage-induced phosphorylation of the Ser-15 residue of p53.

Inhibition of p53 phosphorylation by PI3 kinase inhibitors. (A) HCT116 cells were co-treated with a DNA damaging agent, doxorubicin (200 nM), and either caffeine or wortmannin for 24 h. (B) Cells were co-treated with PTX-2 (100 ng/ml) and the indicated dose of caffeine or wortmannin for 72 h. (C) Cells were co-treated with PTX-2 (100 ng/ml) and the indicated dose of PD98059 or SB202190 for 72 h. Cell extracts were subjected to Western blot analysis with the indicated antibodies.
Since both Ser-15 and Ser-37 of p53 are phosphorylated by ATM kinase in response to DNA damage, ATM-positive and -deficient cells were examined for PTX-2 dependent phosphorylation of those residues of p53. AG04405 cells, which are derived from an AT patient deficient in ATM, and normal fibroblasts such as HDF, WI38, and IMR-90, exhibited cell cycle arrest and phosphorylation of p53 protein upon actin damage (Figures 3A and 3B), indicating that actin damage induces phosphorylation of p53 Ser-15 in an ATM-independent manner. Therefore these results suggest that upstream signaling pathway leading p53 phosphorylation is different in between DNA and actin damage responses.

p53 phosphorylation in ATM-knockout cells. (A) Flow cytometric analysis of human normal fibroblasts HDF and WI38, and ATM-knockout fibroblasts AG04405 after treatment with PTX-2 (100 ng/ml) for 72 h. (B) Cell extracts were prepared from cells treated as in (A) and subjected to Western blot analysis with the indicated antibodies.
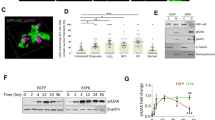
Among PI3 kinases, DNA-PKcs is the most resistant to inhibition by caffeine (Sarkaria et al., 1999). Therefore, we further examined the role of DNA-PKcs in p53 phosphorylation. Induction of p53 expression and Ser-15 phosphorylation was not affected by the knockdown of DNA-PKcs (Figure 4A). However, unexpectedly, DNA-PKcs knockdown cells exhibited enhanced apoptotic death following treatment with PTX-2 in association with an increased number of polyploid cells (Figure 4B). Therefore, these results suggest that DNA-PKcs does not account for p53 phosphorylation, but is involved in the maintenance of tetraploid G1 arrest following actin damage.

p53 phosphorylation and apoptotic death in DNA-PKcs knockdown cells. (A) HCT116 cells were transfected with siRNA targeting DNA-PKcs or GFP as a control. Cells were treated with PTX-2 2 days after siRNA transfection. Cell extracts were subjected to Western blot analysis with the indicated antibodies. (B) Flow cytometric analysis of HCT116 cells transfected with GFP siRNA or DNA-PKcs siRNA after treatment with PTX-2 (100 ng/ml) for 72 h.
Because of p53-independent function of DNA-PKcs in tetraploid G1 arrest, we further examined whether p53 induction is a result of actin damage or polyploidy. To end this, cells were first arrested at early S phase by double thymidine block (Park et al., 2000). While cells treated with PTX-2 alone exhibited tetraploid (4N) and polyploidy (8N), those synchronized by double thymidine block kept arresting at G1/S after the PTX-2 treatment (Figure 5A). However, p53 induction was not observed in cells synchronized by the thymidine block upon treatment with PTX-2 (Figure 5B), implying that the perturbation of actin cytoskeleton by PTX-2 does not induce p53 in cells arrested at G1/S phase. Therefore, these results suggest that p53 induction upon actin damage is not merely a result of perturbation of the actin cytoskeleton. Instead, cell may recognize actin damage-induced polyploidy as a stress signal inducing p53. Taken together these, p53 and DNA-PKcs function independently to sustain tetraploid G1 arrest and prevent polyploidy formation.

PTX-2 does not induce p53 expression in G1/S-arrested cells. (A) HCT116 cells were treated with PTX-2 (100 ng/ml) for 72 h under conditions of normal growth or G1-synchronization by treatment with thymidine as described in Methods. DNA content was analyzed by flow cytometry. (B) Western blot analysis of HCT116 cells treated as in (A).
Methods
Cell culture
Human colorectal cancer cell line HCT116, ATM-deficient fibroblast cell line AG04405, and human normal fibroblast cell lines IMR90 and WI38 were maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum (Life Technologies, Inc., USA) and penicillin-streptomycin (50 U/ml). For knockdown of DNA-PKcs expression, HCT116 cells were transfected with siRNA targeting DNA-PKcs, 5'-AAAGGGCCAAGCTGTCACTCT-3' (Dharmacon Research) using Lipofectamine 2000 (Invitrogen).
Cell cycle synchronization at G1/S of the cell cycle
HCT116 cells were synchronized at G1/S with a modified double thymidine block protocol as previously described (Park et al., 2000). Cells were plated at 0.3 × 106 cells/100-mm dish and 0.1 × 106 cells / 60-mm dish. After 1 day, cells were arrested by treatment with thymidine (5 mM, Sigma Corp.) for 17 h, and were then released from the arrest by washing in Dulbecco's phosphate buffered saline (8 mg/ml NaCl, 0.2 mg/ml KCl, 0.1 mg/ml CaCl2, 0.1 mg/ml MgCl2, 2.31 mg/ml Na2HPO4·12H2O, 0.2 mg/ml KH2PO4) and replenishing with thymidine-free medium. After 9 h, cells were subjected to a second thymidine treatment.
Cell cycle analysis
For DNA content analysis, 1 × 106 cells were harvested by trypsinization and fixed by rapid submersion in 1 ml cold 70% ethanol. After fixation at -20℃ for at least 1 h, cells were pelleted and resuspended in 1 ml staining solution (50 µg/ml propidium iodide, 50 µg/ml RNase, 0.1% Triton X-100 in citrate buffer, pH 7.8), and analyzed with FACScan (Becton Dickinson) using Lysys software (Chae et al., 2004; Jung et al., 2004).
Western blot analyses
Twenty micrograms of protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to PolyScreen membranes (NEN). The membranes were subsequently blocked with 5% nonfat dry milk in Tris-buffered saline Tween20 buffer (Jung et al., 2001) and probed with antibodies. Primary antibodies were detected with a horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or donkey anti-goat secondary antibodies with enhanced chemiluminescence detection (Amersham, UK). Primary antibodies used were followings: anti-p53, anti-p-ser15, anti-p-ser20, anti-p-ser37, anti-actin, anti-p21, anti-DNA-PKcs, (Santa Cruz Biotechnology, CA); and α-tubulin (Sigma-Aldrich, MO).
Abbreviations
- ATM:
-
ataxia telangiectasia mutated protein
- DCB:
-
dihydrocytochalasin B
- DNA-PKcs:
-
DNA-dependent protein kinase catalytic subunit
- PTX-2:
-
pectenotoxin-2
References
Amon A . The spindle checkpoint . Curr Opin Genet Dev 1999 ; 9 : 69 - 75
Andreassen PR, Lohez OD, Lacroix FB, Margolis RL . Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1 . Mol Biol Cell 2001 ; 12 : 1315 - 1328
Ashcroft M, Kubbutat MH, Vousden KH . Regulation of p53 Function and Stability by Phosphorylation . Mol Cell Biol 1999 ; 19 : 1751 - 1758
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B . Requirement for p53 and p21 to sustain G2 arrest after DNA damage . Science 1998 ; 282 : 1497 - 1501
Chae HD, Yun J, Bang YJ, Shin DY . Cdk2-dependent phosphorylation of the NF-Y transcription factor is essential for the expression of the cell cycle-regulatory genes and cell cycle G1/S and G2/M transitions . Oncogene 2004 ; 23 : 4084 - 4088
Chae HD, Choi TS, Kim BM, Jung JH, Bang YJ, Shin DY . Oocyte-based screening of cytokinesis inhibitors and identification of pectenotoxin-2 that induces Bim/Bax-mediated apoptosis in p53-deficient tumors . Oncogene 2005 ; 24 : 4813 - 4819
Chae HD, Kim BM, Yun UJ, Shin DY . Deregulation of Cdk2 causes Bim-mediated apoptosis in p53-deficient tumors following actin damage . Oncogene 2008 ; 27 : 4115 - 4121
Giaccia AJ, Kastan MB . The complexity of p53 modulation: emerging patterns from divergent signals . Genes Dev 1998 ; 12 : 2973 - 2983
Hartwell LH, Weinert TA . Checkpoints: controls that ensure the order of cell cycle events . Science 1989 ; 246 : 629 - 634
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers . Science 1991 ; 253 : 49 - 53
Jordan M, Thrower D, Wilson L . Mechanism of inhibition of cell proliferation by Vinca alkaloids . Cancer Res 1991 ; 51 : 2212 - 2222
Jordan M, Wendell K, Gardiner S, Derry W, Copp H, Wilson L . Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death . Cancer Res 1996 ; 56 : 816 - 825
Jung MS, Yun J, Chae HD, Kim JM, Kim SC, Choi TS, Shin DY . p53 and its homologues, p63 and p73, induce a replicative senescence through inactivation of NF-Y transcription factor . Oncogene 2001 ; 20 : 5818 - 5825
Jung MS, Jin DH, Chae HD, Kang S, Kim SC, Bang YJ, Choi TS, Choi KS, Shin DY . Bcl-xL and E1B-19K proteins inhibit p53-induced irreversible growth arrest and senescence by preventing reactive oxygen species-dependent p38 activation . J Biol Chem 2004 ; 279 : 17765 - 17771
Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, FrankAR, Zhou Y, Bryant MF, Zhou T, Luche DD, Nikolaishvili-Feinberg N, Simpson DA, Cordeiro-Stone M . Caffeine and human DNA metabolism: the magic and the mystery . Mutat Res 2003 ; 532 : 85 - 102
Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB . Wild-type p53 is a cell cycle checkpoint determinant following irradiation . Proc Natl Acad Sci USA 1992 ; 89 : 7491 - 7495
Lanni JS, Jacks T . Characterization of the p53-dependent postmitotic checkpoint following spindle disruption . Mol Cell Biol 1998 ; 18 : 1055 - 1064
Margolis RL . Tetraploidy and tumor development . Cancer Cell 2005 ; 8 : 353 - 354
Margolis RL, Lohez OD, Andreassen PR . G1 tetraploidy checkpoint and the suppression of tumorigenesis . J Cell Biochem 2003 ; 88 : 673 - 683
Minn AJ, Boise LH, Thompson CB . Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage . Genes Dev 1996 ; 10 : 2621 - 2631
Oren M . Regulation of the p53 tumor suppressor protein . J Biol Chem 1999 ; 274 : 36031 - 36034
Park M, Chae HD, Yun J, Jung M, Kim YS, Kim SH, Han MH, Shin DY . Constitutive activation of cyclin B1-associated cdc2 kinase overrides p53-mediated G2-M arrest . Cancer Res 2000 ; 60 : 542 - 545
Paulovich AG, Toczyski DP, Hartwell LH . When checkpoints fail . Cell 1997 ; 88 : 315 - 321
Rudner AD, Murray AW . The spindle assembly checkpoint . Curr Opin Cell Biol 1996 ; 8 : 773 - 780
Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT . Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine . Cancer Res 1999 ; 59 : 4375 - 4382
Stewart ZA, Pietenpol JA . Cell cycle checkpoints as therapeutic targets . J Mammary Gland Biol Neoplasia 1999 ; 4 : 389 - 400
Torres K, Horwitz SB . Mechanisms of Taxol-induced cell death are concentration dependent . Cancer Res 1998 ; 58 : 3620 - 3626
Ui M, Okada T, Hazeki K, Hazeki O . Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase . Trends Biochem Sci 1995 ; 20 : 303 - 307
Acknowledgements
This work was supported by the Institute of Bioscience and Technology (IBST) of Dankook university and by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-359-C00030).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chae, HD., Kim, S., Park, S. et al. p53 and DNA-dependent protein kinase catalytic subunit independently function in regulating actin damage-induced tetraploid G1 arrest. Exp Mol Med 44, 236–240 (2012). https://doi.org/10.3858/emm.2012.44.3.018
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2012.44.3.018
