
Abstract
Yunis-Varón syndrome (YVS) is an autosomal recessive disorder comprising skeletal anomalies, dysmorphism, global developmental delay and intracytoplasmic vacuolation in brain and other tissues. All hitherto-reported pathogenic variants affect FIG4, a lipid phosphatase involved in phosphatidylinositol (3,5)-bisphosphate [PtdIns(3,5)P2] metabolism. FIG4 interacts with PIKfyve, a lipid kinase, via the adapter protein VAC14; all subunits of the resulting complex are essential for PtdIns(3,5)P2 synthesis in the endolysosomal membrane compartment. Here, we present the case of a female neonate with clinical features of YVS and normal FIG4 sequencing; exome sequencing identified biallelic rare coding variants in VAC14. Cultured patient fibroblasts exhibited a YVS-like vacuolation phenotype ameliorated in a dose-dependent fashion by ML-SA1, a pharmacological activator of the lysosomal PtdIns(3,5)P2 effector TRPML1. The patient developed a diffuse leukoencephalopathy with loss of the normal N-acetylaspartate spectrographic peak and presence of a large abnormal peak consistent with myoinositol. We report that VAC14 is a second gene for Yunis-Varón syndrome.
Similar content being viewed by others


Introduction
Yunis-Varón syndrome (YVS; OMIM #216340) is an autosomal recessive syndrome characterized by skeletal anomalies (cleidocranial dysplasia; absence/hypoplasia of thumbs, halluces and distal phalanges; gracile bones; hypomineralization and fractures), brain malformations, dysmorphic features and poor outcome.1, 2 All pathogenic variants thus-far reported affect FIG4, a lipid phosphatase involved in phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2) metabolism.3 PtdIns(3,5)P2 is enriched at the cytosolic surface of endolysosomal membranes where it is synthesized by a ternary complex consisting of FIG4, the lipid kinase PIKfyve and the adapter protein VAC14; all three proteins are essential for PtdIns(3,5)P2 synthesis.4, 5 Cells from YVS patients and orthologous (Fig4-/-) mice have altered endolysosomal trafficking, as evidenced by the presence of multiple, enlarged vacuoles expressing endolysosomal markers including LAMP2.3, 6, 7
Pathogenic variants of the PtdIns(3,5)P2 synthetic complex cause several disorders. The clinical spectrum of FIG4 includes YVS, bilateral temporo-occipital pachygyria (OMIM #612691),8 Charcot-Marie-Tooth disease type 4J (OMIM #611228),7 and autosomal dominant amyotrophic lateral sclerosis (OMIM #612577).9 Haploinsufficiency of PIKFYVE causes François-Neetens fleck corneal dystrophy (OMIM #121850).10 Recently, biallelic VAC14 variants have been described in two probands with childhood-onset striatonigral degeneration (#617054), a neurological condition with onset beyond age 18 months.11 Here, we present a neonate with YVS and biallelic loss-of-function variants in VAC14, the first such case reported to date.
Materials and methods
All procedures were in accord with the Declaration of Helsinki and informed consent was obtained from all participants. The study was approved by the Children's Hospital of Eastern Ontario Research Ethics Board. Exome sequencing was performed on a clinical basis in a commercial laboratory (GeneDx, Maryland, USA) and data were reanalyzed using our in-house annotation pipeline (Supplementary Methods). Fibroblast cultures were established from a 2 mm skin punch biopsy; enzymological studies and filipin staining in fibroblasts were performed in a diagnostic laboratory (AMC Amsterdam) according to standard protocols. For electron microscopy, fibroblasts were trypsinized and fixed in glutaraldehyde solution, after which resin-embedded 90 nm cell sections were stained with osmium tetroxide, uranyl acetate and lead citrate and imaged according to standard protocols. For ML-SA1 rescue experiments, fibroblasts were incubated with vehicle (DMSO) containing 0 μM, 40 μM or 80 μM ML-SA1 (Sigma) for 24 h, rinsed in PBS and imaged directly.
Results
Clinical findings
The proband is the female first child of healthy nonconsanguineous parents. The pregnancy was notable for intrauterine growth restriction during the third trimester, and a term Cesarean delivery was performed due to fetal distress. The child was small for gestation (weight=2300 g (Z=−2.25); head circumference=32cm (Z=−1.8); length=46cm (Z=−1.44)), hypotonic and dysmorphic (Figures 1a–f). Skeletal films (Figures 1g–m) showed diffuse osteopenia, gracile long bones, multiple diaphyseal fractures, ‘handlebar’ clavicles and hypoplasia of thumbs, halluces and distal phalanges. Cranial ultrasound and serial cranial MRI showed several striking abnormalities, including bilateral cystic basal ganglial degeneration, a progressive leukodystrophy and MR spectrographic changes consistent with gliosis (Figure 2). Reflexes were absent; nerve conduction studies showed an axonal polyneuropathy. The patient was given a clinical diagnosis of YVS. Her course has been characterized by failure to thrive, profound developmental delay and hydrocephalus necessitating shunt placement at age seven months. Now eight months of age, she is unreactive with persistent ophisthotonus, roving eye movements, severe feeding intolerance and episodes of increased irritability.

Examination shortly after birth (a,d) was notable for large anterior fontanelle, low-set ears, bilateral cortical cataracts, short nose with anteverted nares, micrognathia and short halluces. By five months of age (b,c,e,f) she had developed the additional findings of sparse hair, macrocephaly (Z=+5) and a tall, bossed, prominent forehead. Neonatal skeletal survey (g–m) showed osteopenia, permeative appearance of bone, gracile long bones, ‘handlebar’ clavicles, flared iliac wings, acetabular dysplasia, vertebral disc calcifications and hypoplasia of multiple bones in the hands and feet (first metacarpals and metatarsals, all distal phalanges, proximal phalanges of the thumbs and middle phalanges of fifth fingers). Also seen were healing fractures of the distal femora and proximal tibiae, and periosteal new bone formation of the long bones. Lumbar spine bone mineral density (BMD) was reduced (Z=−1.9). Pamidronate treatment improved the patient’s tachypnea, analgesia requirements and bone mineral density (Z=−0.25 at age five months). Shown are (g) anterio-posterior supine film; (h) right humerus; (i) left radius and ulna; (j) left hand; (k) left femur; (l) left tibia and fibula; (m) right foot.

Serial brain MRI at the ages of 6 days (a–c), 6 weeks (d–f) and 8.5 months (g–i) showed progressive cystic degeneration of the basal ganglia and a diffuse, progressive leukoencephalopathy. The initial imaging finding was hyperechogenicity and thickening of the frontal subcortical white matter on a cranial ultrasound at two days of age (j). MRI showed corresponding symmetric T2-hyperintense and T1-hypointense signal changes (c), which progressed rapidly by 8.5 months to involve all white matter areas in a confluent manner (i). The overlying cortical sulci have taken on a flattened appearance (g–i). MR spectroscopy of the centrum semiovale at 8.5 months (k) showed a smaller-than-normal N-acetylaspartate (NAA) peak, in conjunction with an abnormally large peak at 3.5 PPM, the chemical shift of myoinositol. The observed spectrum is consistent with disrupted neuronal integrity and gliosis. Diffusion-weighted images were normal (data not shown).
Genetic investigations
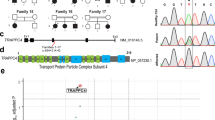
FIG4 sequencing and genomic SNP microarray in the proband were normal. Clinical whole-exome sequencing of the proband and parents identified biallelic variants in VAC14 [NM_018052.3]. The changes are a paternally-inherited substitution, c.1895C>T (p.Thr632Met), and a maternally-inherited stop mutation, c.923T>A (p.Leu308*); both are rare and predicted to be deleterious (Supplementary Table 1). We re-annotated and reviewed all rare, nonsynonymous coding (and copy-number) variants consistent with de novo dominant, homozygous, or compound heterozygous segregation (Supplementary Methods); the resulting list of 10 genes (Supplementary Table 1) contains no other candidates likely to account for the patient’s phenotype.
Biochemical investigations
We assessed fibroblasts from the proband for evidence of disordered endolysosomal trafficking, as follows: Microscopic examination of patient fibroblasts confirmed the presence of numerous, swollen cytoplasmic vacuoles with an ‘empty’ ultrastructural appearance, as described in YVS (Figure 3).3, 6 Vacuolation was partially rescued by ML-SA1, a pharmacological activator of the (PtdIns(3,5)P2-gated) transient receptor potential channel TRPML1.12 We measured the in vitro activities of several lysosomal acid hydrolases in serum and patient fibroblasts (Supplementary Table 2), noting a slight, persistent increase in serum total beta-hexosaminidase activity. Arylsulphatase A activity was slightly reduced in patient fibroblasts (no variants were detected in ARSA, all coding portions of which were sequenced >10-fold); the activities of all other lysosomal enzymes tested were within normal limits.

Prominent intracytoplasmic vacuoles were observed in a subset of patient-derived fibroblasts (a,c) but not in control fibroblasts (b,d). (a,b) phase-contrast microscopy (scale bar=100 μm). (c,d) electron microscopy (scale bar=2 μm): vacuoles have an ‘empty’ ultrastructural appearance and are delimited by a single membrane. Similar (less obvious) findings were also observed in positive control fibroblasts from a patient with FIG4-related YVS (data not shown). (e–h) Treatment of cells with a pharmacological TRPML1 activator (ML-SA1) resulted in dose-dependent rescue. Patient cells (treated with 0, 40, or 80 μmol/l ML-SA1 for 24 h) were scored as containing ‘large vacuoles’ (e), ‘small vacuoles’ (f), or ‘no vacuoles’ (g). >125 cells were scored for each condition in each of three biological replicates; two-way ANOVA (h) showed a significant (*P<0.05; ***P<0.001) increase in cells with ‘no vacuoles’ after treatment with 80 μmol/l ML-SA1.
During the patient’s initial clinical assessment, several features (large anterior fontanelle, leukodystrophy, osteopenia and fractures) prompted assessment of peroxisomal functions (Supplementary Table 2). Interestingly, three distinct markers of peroxisomal beta-oxidation of very long chain fatty acids [VLCFAs] (VLCFAs, C26:0 lysophosphatidylcholine levels, and oxidation of labeled C22:0 in fibroblasts) were subtly abnormal, although VLCFAs in blood showed no consistent abnormality. No differences in the activity or expression of any of the enzymes directly involved in peroxisomal beta-oxidation were observed directly.
Discussion
We report here for the first time that pathogenic variants of VAC14 are a cause of YVS. The features shared by our proband and the described individuals with FIG4-related YVS (Supplementary Table 3) include dysmorphism, skeletal (thumb/halluceal, phalangeal and clavicular) dysplasia, and, in surviving patients, severe global developmental delay. Osteopenia, congenital fractures, and gracile long bones, although less-frequently reported, are described components of the skeletal phenotype.2, 13 The spectrum of described neurological manifestations in YVS is very broad, including pachygyria, polymicrogyria, callosal and/or cerebellar vermis hypoplasia, Dandy-Walker malformation, ventriculomegaly, hydrocephalus, and white matter atrophy.2, 6, 13, 14 As far as we are aware, the MRI and magnetic resonance spectroscopy findings in our patient (leukodystrophy, spectroscopic changes, and cystic basal ganglial degeneration) are previously-unreported, comparable clinical reports being few. It remains to be seen whether the latter findings are VAC14-specific, or a general feature of YVS irrespective of genotype.
We are aware of at least two well-characterized mouse models of VAC14-related neurodegeneration. Constitutive Vac14-null mice have a perinatal-lethal vacuolar neurodegeneration phenotype with deficient recycling of the cation-independent mannose-6-phosphate receptor (M6PR), the M6PR being the mechanism whereby acid hydrolases are targeted to the lysosome.15 The second model, ingls (infantile gliosis), is caused by a recessive Vac14 substitution, and is characterized by small size, hypopigmentation, motor impairments, spongiform degeneration of the thalamus, brainstem, and cerebellar nuclei, and lethality by age three weeks.4 Abnormal intracellular vacuolation is a described feature of both models.
The precise cellular trafficking defect(s) in YVS remain to be defined. PtdIns(3,5)P2 signaling has been implicated in diverse endolysosomal transactions, including lysosomal charge/pH regulation, autophagosome-lysosome fusion, multivesicular body cargo selection, and endosome-to-trans-Golgi network retrograde transport.16 In this study, vacuolation was partially suppressed by ML-SA1, a pharmacological activator of TRPML1/mucolipin-1. The latter protein is a PtdIns(3,5)P2-gated calcium channel proposed to regulate lysosomal fission.17 Considering the multiple cellular roles of PtdIns(3,5)P2, other, potentially multiple, pathways are likely to be involved. For example, the patient’s persistently increased serum hexosaminidase activity is suggestive of a partial defect of M6PR-mediated lysosomal enzyme targeting, as described already in murine, Drosophila, and HeLa models of YVS.15, 18, 19
The apparent partial defect of peroxisomal beta-oxidation of VLCFAs in our patient, which was consistent across three different methods, is currently unexplained. Recently, lysosome-peroxisome contacts mediated by another PI, phosphoinositol 4,5-bisphosphate, have been described in the context of intracellular cholesterol transport;20 an analogous pathway is not yet described for PtdIns(3,5)P2. Detailed biochemical observations in additional cases will be needed to clarify the mechanistic basis of this rare and devastating illness.
References
Yunis E, Varón H : Cleidocranial dysostosis, severe micrognathism, bilateral absence of thumbs and first metatarsal bone, and distal aphalangia: a new genetic syndrome. Am J Dis Child 1980; 134: 649–653.
Basel-Vanagaite L, Kornreich L, Schiller O, Yacobovich J, Merlob P : Yunis-Varon syndrome: further delineation of the phenotype. Am J Med Genet A 2008; 146A: 532–537.
Campeau PM, Lenk GM, Lu JT et al: Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013; 92: 781–791.
Jin N, Chow CY, Liu L et al: VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J 2008; 27: 3221–3234.
Bharadwaj R, Cunningham KM, Zhang K, Lloyd TE : FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum Mol Genet 2016; 25: 681–692.
Walch E, Schmidt M, Brenner RE et al: Yunis-Varon syndrome: evidence for a lysosomal storage disease. Am J Med Genet 2000; 95: 157–160.
Chow CY, Zhang Y, Dowling JJ et al: Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007; 448: 68–72.
Baulac S, Lenk GM, Dufresnois B et al: Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology 2014; 82: 1068–1075.
Chow CY, Landers JE, Bergren SK et al: Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 2009; 84: 85–88.
Li S, Tiab L, Jiao X et al: Mutations in PIP5K3 are associated with François-Neetens mouchetée fleck corneal dystrophy. Am J Hum Genet 2005; 77: 54–63.
Lenk GM, Szymanska K, Debska-Vielhaber G et al: Biallelic mutations of VAC14 in pediatric-onset neurological disease. Am J Hum Genet 2016; 99: 188–194.
Zou J, Hu B, Arpag S et al: Reactivation of lysosomal Ca2+ efflux rescues abnormal lysosomal storage in FIG4-deficient cells. J Neurosci 2015; 35: 6801–6812.
Garrett C, Berry AC, Simpson RH, Hall CM : Yunis-Varon syndrome with severe osteodysplasty. J Med Genet 1990; 27: 114–1121.
Nakajima J, Okamoto N, Shiraishi J et al: Novel FIG4 mutations in Yunis-Varon syndrome. J Hum Genet 2013; 58: 822–824.
Zhang Y, Zolov SN, Chow CY et al: Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci USA 2007; 104: 17518–17523.
Jin N, Lang MJ, Weisman LS : Phosphatidylinositol 3,5-bisphosphate: regulation of cellular events in space and time (Review). Biochem Soc Trans 2016; 44: 177–184.
Dong XP, Shen D, Wang X et al: PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 2010; 1: 38.
de Lartigue J, Polson H, Feldman M et al: PIKfyve regulation of endosome-linked pathways. Traffic 2009; 10: 883–893.
Rutherford AC, Traer C, Wassmer T et al: The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci 2006; 119: 3944–3957.
Chu BB, Liao YC, Qi W et al: Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 2015; 161: 291–306.
Acknowledgements
The authors gratefully acknowledge the patient and family for their contribution. This work was performed under the Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec, and Children’s Hospital of Eastern Ontario Foundation.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Lines, M., Ito, Y., Kernohan, K. et al. Yunis-Varón syndrome caused by biallelic VAC14 mutations. Eur J Hum Genet 25, 1049–1054 (2017). https://doi.org/10.1038/ejhg.2017.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.99
This article is cited by
-
A new UHPLC-MS/MS method for the screening of urinary oligosaccharides expands the detection of storage disorders
Orphanet Journal of Rare Diseases (2021)
-
A dysfunctional endolysosomal pathway common to two sub-types of demyelinating Charcot–Marie–Tooth disease
Acta Neuropathologica Communications (2020)
-
Homozygous variant, p.(Arg643Trp) in VAC14 causes striatonigral degeneration: report of a novel variant and review of VAC14-related disorders
Journal of Human Genetics (2019)
