
Abstract
Twelve individuals of consanguineous Bedouin kindred presented with autosomal recessive progressive spastic paraplegia evident as of age 0–24 months, with spasticity of lower limbs, hyperreflexia, toe walking and equinus deformity. Kyphoscolisois was evident in older patients. Most had atrophy of the lateral aspects of the tongue and few had intellectual disability. Nerve conduction velocity, electromyography and head and spinal cord magnetic resonance imaging were normal in tested subjects. Muscle biopsy showed occasional central nuclei and fiber size variability with small angular fibers. Genome-wide linkage analysis identified a 6.7Mbp disease-associated locus on chromosome 3q21.3–3q22.2 (LOD score 9.02; D3S1290). Whole-exome sequencing identified a single homozygous variant within this locus, c.51_52ins(28); p.(V18fs56*) in KY, segregating in the family as expected and not found in 190 Bedouin controls. High KY transcript levels were demonstrated in muscular organs with lower expression in the CNS. The phenotype is reminiscent of kyphoscoliosis seen in Ky null mice. Two recent studies done independently and parallel to ours describe somewhat similar phenotypes in one and two patients with KY mutations. KY encodes a tranglutaminase-like peptidase, which interacts with muscle cytoskeletal proteins and is part of a Z-band protein complex, suggesting the disease mechanism may resemble myofibrillar myopathy. However, the mixed myopathic–neurologic features caused by human and mouse Ky mutations are difficult to explain by loss of KY sarcomere stabilizing function alone. KY transcription in CNS tissues may imply that it also has a role in neuromotor function, in line with the irregularity of neuromuscular junction in Ky null mutant mice.
Similar content being viewed by others

Introduction
Hereditary spastic paraplegia (HSP) denotes a large and diverse group of neurodevelopmental or neurodegenerative disorders that vary in genetic etiology, pathophysiology and clinical manifestations. The main attribute of HSP is weakness and spasticity of both lower extremities in various degrees. Symptoms may present at any stage of life, from birth to late adulthood, with varying rates of progression. Patients with HSP display increased muscle tone in some muscles of the lower extremities with weakness in others, hyperreflexia, occasional reduced vibration sensation and spastic gait. HSP may be limited to the lower extremities and urinary bladder, or it may be complicated by various neurologic deficits, such as intellectual disability (eg, SPG54; MIM #615003), brain abnormalities (eg, SPG11; MIM #610844) and even vitiligo (SPG 23; MIM #270750).1, 2 Phenotypic variability is sometimes present within distinct genetic entities, as has been demonstrated in CYP7B1 mutations (SPG5A; MIM #603711), where optic atrophy, nystagmus and cerebellar signs are found in only some of the patients.
The most common known pathophysiology of HSP is axonopathy of the long motor fibers of the corticospinal tract to the lower extremities, generated through a wide array of molecular mechanisms.2 The genetic heterogeneity of HSP syndromes is vast: there are over 70 genetic subtypes recognized to date, with various modes of inheritance, and many of the genetic variants are yet to be unraveled. Molecular genetic diagnosis of HSP is further complicated by the great clinical overlap between syndromes and by the similarity of HSP to other clinical entities2, 3 We now describe a novel homozygous loss-of-function mutation in KY (#MIM 617114), causing a form of HSP in 12 individuals in an Israeli Bedouin kindred.
Materials and methods
Patients
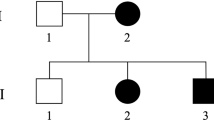
Twelve affected individuals (ages newborn to 52 years) of three different families of a single consanguineous Bedouin Israeli tribe were studied (Figure 1a). DNA and RNA samples were obtained following Soroka Medical Center IRB approval and informed consent. Clinical phenotyping was determined by experienced pediatric neurologists, orthopedic surgeon and geneticist for all affected individuals. Seven patients underwent either computed tomography (CT) or magnetic resonance imaging (MRI) of the head, spine or both. One patient was tested for electromyography (EMG) and nerve conduction velocity (NCV). According to patients’ accounts, there are at least 10 more individuals in the extended family who suffer from the same phenotype, but were either unavailable or declined to participate in the study.

Pedigrees and phenotype: (a) pedigrees of three affected families of the same Bedouin tribe from the Negev area of Israel. (b) X-ray imaging showing tip-toe walking position and skeletal deformity of the foot of patient P.1 IV:3 at age 2 years. (c) X-ray and MRI of patient P.1 IV:12, age 13 years, showing thoracic scoliosis and kyphosis. (d) Hematoxylin and eosin staining of gastrocnemius muscle of patient P.1 IV:3, age 3 years, showing occasional central nuclei (black arrows), small, angular fibers (blue arrows) and fiber size variability. The full colour version of this figure is available at European Journal of Human Genetics online.
Muscle pathology
Following informed consent, muscle tissue of the gastrocnemius muscle was sampled bilaterally from a 3-year-old patient during orthopedic surgery. Muscle specimens were divided, and samples were formalin fixed for histological studies and snap-frozen for RNA extraction. RNA was extracted using Genzol reagent (Geneaid, New Taipei City, Taiwan) and reverse-transcribed using Verso cDNA kit (Thermo Scientific, Waltham, MA, USA). Fixed tissues were paraffin embedded, sectioned and stained with H&E using standard techniques.
Sequencing
Whole-exome sequencing (HiSeq2500, Illumina, San Diego, CA, USA) of individual P.2 II:3 (Figure 1a) was performed using paired-end 100-bp read protocol at a mean coverage of 70-fold (96.7% of all exonic nucleotides and splice junctions were covered by >10 reads). For exome enrichment, we used SureSelect Human All Exon V5+UTR enrichment kit (Agilent, Santa Clara, CA, USA). Sequencing read alignment, variant calling and annotation were performed by Macrogen (Seoul, Korea) using BWA, GATK and SnpEff, respectively. Data were analyzed through the use of QIAGEN’s Ingenuity Variant Analysis software (www.qiagen.com/ingenuity, QIAGEN, Redwood City, CA, USA). Using the filtering cascade, we excluded variants that were observed with an allele frequency ≥0.5% of the genomes in the 1000 genomes project, NHLBI ESP exomes (All), ExAC data set or the Allele Frequency Community. In addition, we excluded variants which appeared in a homozygous state in our in-house whole-exome sequencing database of 200 Bedouin control samples. We then kept variants which were predicted to have a deleterious effect on protein coding sequences (eg, Frameshift, in-frame indel, stop codon change, missense or predicted to disrupt splicing by MaxEnt Scan), and variants which were experimentally observed to be associated with a phenotype: pathogenic, possibly pathogenic, uncertain significance or disease-associated according to the Human Gene Mutation Database, or have been established or predicted to have a gain-of-function effect. Variants were separately filtered for genes known to cause HSP and neuromuscular phenotypes.
Linkage analysis
Genome-wide linkage analysis of four affected individuals (P.1 IV:2 and IV:11, P.2 II:3 and P.3 IV:1; Figure 1a) was performed using Illumina Omni Express Beadchip with >700 K SNP loci per sample (Illumina, San Diego, CA, USA). Homozygosity mapping analysis was carried out using the open online software Homozygosity-Mapper4 assuming equal allele frequencies with no lower threshold for size of homozygosity loci (Figure 2a). Haplotype reconstruction of the chromosome 3 locus, testing all available samples of the kindred, was performed using polymorphic markers D3S1587, D3S1273, D3S1576 and D3S1290 as previously described.5 All physical positions mentioned are according to the GRCh37/hg19 genome assembly. Multipoint LOD score at the shared locus was calculated using SUPERLINK ONLINE SNP 1.1,6 assuming an autosomal recessive mode of inheritance with penetrance of 0.99 and disease mutant gene frequency of 0.01.

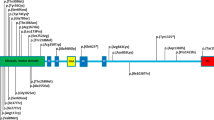
The KY mutation and expression pattern: (a) homozygosity mapping showing a shared homozygosity locus in chromosome 3 in four affected individuals (P.1 IV:2, IV:11, P.2 II:3 and P.3 IV:1). (b) Sanger sequencing of KY amplicon of an unaffected individual (P.1V:2), an obligatory carrier (P.3 III:1), and an affected individual (P.3 IV:1). Insertion site marked by red rectangle. (c) RT-PCR of normal human tissues demonstrating the tissue specificity of KY (GAPDH as positive control; genomic DNA as negative control not shown). (d) Schematic representation of KY protein with known human (red) and murine (blue) variations; TGc, transglutaminase domain. The full colour version of this figure is available at European Journal of Human Genetics online.
Validation and segregation analysis
Validation and segregation analysis of the KY variant (Clinvar accession number SCV000494644) was done using two techniques: Sanger sequencing and restriction fragment length polymorphism (RFLP) analysis. PCR primers sequences used for both validations were: forward 5′-TAATGGCGTGTTTGGCAGAG-3′; reverse 5′-CCCTTGTGGCAAGGAGG-3′. For RFLP, the KY c.51_52ins(28) variant introduces an AflIII restriction site: restriction analysis of the 405-bp mutant allele amplicon yields two fragments (248 and 157 bp), whereas the single 377-bp wild-type allele amplicon remains uncut.
KY expression analysis
A panel of cDNA samples was prepared from total RNA derived from 20 normal human tissues (Clontech Laboratories, Mountain View, CA, USA), as well as Genezol extracted RNA of PBMC and EBV-transformed lymphocytes from an ethnically matched individual, using Verso cDNA kit (Thermo Scientific). Genomic DNA control from an ethnically matched individual was prepared using standard methods. Two sets of PCR primers were designed to amplify cDNA rather than genomic DNA of human KY and of glyceraldehyde 3 phosphate dehydrogenase (GAPDH) housekeeping gene as a control. The reaction was set for 35 cycles at Tm of 60 °C. Primers used for KY amplification (241- bp amplicon): forward 5′-CCCACCCAGAGACTTCCATG-3′; reverse 5′- TTGAGCGTGTACTCCAGCAC-3′; primers used for GAPDH (452 bp amplicon): forward 5′-ACCACAGTCCATGCCATCAC-3′; reverse 5′-TCCACCACCCTGTTGCTGT-3′.
Quantitative analysis of KY transcript in affected muscle tissue was performed by qPCR using primers: forward 5′- AAAGATGCCCATGCCTACCCC-3′; reverse 5′-CAGGTCACTCACCAGTTCGTC-3′ (138- bp amplicon). Primers used for GAPDH transcript detection: forward 5′-TCGACAGTCAGCCGCATC-3′; reverse 5′-CCGTTGACTCCGACCTTC-3′ (87-bp amplicon).
Results
Twelve individuals of a single consanguineous Bedouin tribe in southern Israel (Figure 1a), born after uneventful pregnancies, were affected with a similar neurological disorder: all patients showed signs of progressive spastic paraplegia with very early onset, evident at 5 days to 2 years of age, most seeking medical attention at an early age because of spasticity, delay in achieving motor developmental milestones or tip-toe walking (Figure 1b). A detailed comparison of phenotypic attributes is presented in Table 1. Tip-toeing was progressive and was exacerbated by walking or running. Neurological exam demonstrated normal cerebellar and cranial nerve function, increased lower extremity muscle tone with brisk hyperactive reflexes but no clonus. Tight Achilles tendon was evident with different levels of equinus deformity, requiring surgical repair in most: most patients went through either Achilles tendon lengthening, gastrocnemius recession or botulinum toxin injection, with mixed results. Some patients exhibited bilateral positive Babinski sign. Spasticity was disabling in some cases, interfering with standing up, and produced a weak positive Gower sign. There was no sensory deficit or urinary dysfunction. Upper extremities showed no signs of abnormal motor or sensory function, apart from atrophy and weakness in two of the patients. Most patients beyond the age of 10 years developed various degrees of either kyphosis, scoliosis or both as demonstrated in patient P.1 IV:12, age 13 years, with mild 'S' shaped thoracic scoliosis and kyphosis of 70 ° (Figure 1c). Several patients showed atrophy of the lateral aspects of the tongue with a groove defining the atrophic and normal parts of the tongue. As shown in Table 1, speech disorders were noted in most patients and various degrees of intellectual disability was evident in some (IQ testing was not performed). Other than the above, there were no dysmorphic features, ophthalmological abnormalities, hearing impairment, cutaneous lesions or any other significant clinical findings associated with the disease.
Creatine kinase levels and metabolic screen were normal. Imaging studies (CT or MRI) of the brain and spinal cord, done for patients ages 1.3 to 21 years, were normal (data not shown). NCV studies of the peroneal and tibial nerves (patient P.1 IV:12 at age 13 years) demonstrated normal distal latencies, amplitudes and velocities. The F responses were normal and sensory studies of the medial plantar nerves showed normal latency, amplitude and velocity. EMG studies of the tibialis anterior and medial gastrocnemius muscles showed no evidence of myopathic or of spontaneous activity or other neurogenic changes. Pathological evaluation of muscle tissues stained with H&E (patient P.1 IV:3, age 3) revealed no signs of necrosis, inflammation, atrophy or sclerosis. Occasional central nuclei and small angular fibers could be observed, as well as variability of muscle fibers size (Figure 1d).
Genome-wide homozygosity analysis of four affected individuals of three distinct related families identified a single 6.7 Mb homozygous segment common to all. The identified segment on chromosome 3q21.3–3q22.2 (Figure 2a) spans between SNPs rs6775459 and rs1502174. Haplotype reconstruction using polymorphic markers D3S1587, D3S1273, D3S1576 and D3S1290, performed for all available DNA samples of the three pedigrees, demonstrated that the genomic segment segregated within the kindred as expected for autosomal recessive heredity with full penetrance. Using all the microsatellite markers mentioned above, the three pedigrees combined yielded a maximum multipoint LOD score of 9.02 at marker D3S1290.
Whole-exome sequencing data of individual P.2 II:3 (Figure 1a) was filtered for known variants (see Materials and methods section), followed by Sanger sequencing of exonic regions with six reads or less within the locus. Following exome data filtering, only a single variant was found within the 6.7 Mbp locus: c.51_52ins (5′-TATCGACATGTGCTGTATCTATCGACAT-3′), hereinafter termed in brief c.51_52ins(28). This homozygous frameshift variant in exon 1 of KY (RefSeq accession number NM_178554.4) is predicted to cause a premature termination of the mature encoded protein p.(V18fs56*). Within the locus, there were no other variants, nor were there any variants in genes known to cause HSP and HSP-like diseases throughout the exome. The KY variant was validated via Sanger sequencing (Figure 2b) and restriction analysis (data not shown), and was found to fully segregate as expected for autosomal recessive heredity in the studied kindred. Of 190 non-related ethnically matched controls of tribes other than the one affected, none carried the variant (data not shown). However, screening of individuals of the same tribe showed high prevalence of carriers (17 heterozygotes of 72 individuals tested, data not shown).
KY expression pattern was analyzed in a cDNA panel of human tissues (RT-PCR), demonstrating high levels of KY transcripts in muscular organs such as skeletal muscle, heart and uterus, with lower expression in the CNS, in peripheral blood and in several organs of the digestive and reproductive systems (Figure 2c). Despite the putative disruptive nature of the c.51_52ins(28) variant and the new premature stop codon, no nonsense mediated decay (NMD) was noted: KY transcript levels in gastrocnemius muscle tissue of an affected individual were similar to those seen in the gastrocnemius of an ethnically matched healthy control sample (data not shown).
Discussion
We describe a novel form of autosomal recessive spastic paraplegia caused by a variant in KY encoding Kyphoscoliosis peptidase. The fact that haplotypes of the 6.7Mb locus were identical between individuals of three distinct families of the same highly inbred tribe implies a common ancestral origin for this mutation. Inactivating mutations in Ky were previously described in mice to cause autosomal recessive kyphoscoliosis and muscle weakness resembling the human disease reported here: the deleterious effect of Ky mutations was first appreciated by the identification of a mouse strain suffering from muscle degeneration in postural, slow contracting muscles, leading to severe thoracolumbar kyphoscoliosis and abnormal gait.7 The causative mutation in mice was identified as a 2-bp deletion in the first exon of KY-ortholog, resulting in a premature stop codon at position 105 of the encoded protein.8 Histological analysis of ky/ky mutant mice showed muscle necrosis in early life, followed by atrophy and fibrosis with mixed characteristics of both neurogenic (eg, axonal sprouting) and myopathic (eg, myofibril necrosis, lack of fiber type grouping) changes. Electron microscopy of ky/ky muscle showed Z-band disruption.9 Despite normal gross microscopical appearance of CNS tissue,9 there was neuromuscular junction (NMJ) disorganization coupled with extra-synaptic expression of AchR, suggesting neural involvement.8, 10
Murine ky has been named so based on the readily evident kyphoscoliosis that developed postnatally in homozygous null mutant mice because of dystrophy of postural muscles. Similar to the mouse phenotype, affected human individuals in our cohort had kyphosis, but it became variably evident only after 10 years of age. The hallmark of the human phenotype was spasticity of the lower limbs. A unique characteristic of the human disease seen in most of our patients was tongue atrophy, restricted to the lateral aspects of the tongue. Other features of the human disease, presenting with variable expression, were scoliosis, developmental delay and muscular weakness (Table 1).
Affected mice and humans differ in terms of muscle tone: affected mice showed diffuse weakness and muscle flaccidity, whereas the human patients showed muscle spasticity, which in conjunction with hyperreflexia suggests combined upper motor neuron and muscle involvement rather than pure muscular pathology. This finding is supported by the EMG studies showing no evidence of myopathic changes. Pathological evaluation showed mild muscle fiber size variation, with occasional central nuclei and small, angular fibers, signs of distinct myopathic and neurological dysfunction11, 12 (Figure 1d). No gross inflammation, necrosis, atrophy or sclerosis were noted in muscle histology (patient P.1.IV:3, age 3 years), in contrast with the necrosis that was evident early on in affected mice.
The murine mutation bears great similarity to the one seen in our patients, both being frameshift variants in the first exon, causing premature stop codon before the predicted catalytic site of the protein (Figure 2d). As deduced by sequence alignment, identity of the murine and human proteins is 90.5% and similarity is 94.7%.13 It is of interest to note that while both the mouse and the human mutations are frameshift mutations in the first exon, normal to high levels of KY mutant transcripts are seen in human muscle, in contrast to those seen in mice.8 It is plausible that part of the phenotypic differences between affected mice and humans result from this difference. Altogether, although some differences exist between the mouse and human phenotypes, the similarity of the phenotypes and genotypes seen in humans and mice, along with the LOD score of 9.02, clearly implicate the KY c.51_52ins(28); p.(V18fs56*) mutation as the cause for HSP in the kindred described.
As delineated above, our patients show signs of a novel progressive neuromuscular disorder, which is in partial concordance with that seen in mice affected by a similar mutation. Two recent homozygosity studies, done in parallel to and independent of our study, identified a c.1071delG KY variant in a single human patient14 and a c.405C>A KY variant in two siblings15 (Figure 2d) The c.1071delG p.(Thr358Leufs*3) loss-of-function variant was described in a 7.5-year-old Kurdish girl with walking difficulties because of muscle weakness and atrophy of the lower limbs and progressive equinus contractures of the feet. She had mild contractures in the shoulders, hips and feet, cavus feet and lordosis but no scoliosis, and had undergone Achilles tendon elongation surgery. Whole-body MRI showed atrophy and fatty infiltration in the calf muscles. The c.405C>A p.(Y135*) substitution variant was demonstrated in two adult Israeli–Arab brothers, with a slowly progressive myopathy from infancy. The lower limbs were primarily affected with muscle weakness and atrophy, whereas upper limb involvement became apparent later. Similar to our patients, affected individuals described in both studies had congenital bilateral equinovarus foot deformity and Achilles tendon contractures that required multiple interventions. Unlike the Kurdish affected individual, yet similar to the mouse phenotype, several of our patients and the Israeli–Arab patients had kyphosis with rigid spine appearing mostly beyond 10 years of age, and some also showed tongue atrophy. Intellectual disability and developmental delay are described in several of both our patients and those with the c.405C>A variant.15
It is of interest to note that the majority of patients described here did not suffer from muscle weakness, as did individuals with the c.405C>A and c.1071delG variants, but rather from spasticity, which was accompanied by hyperreflexia and in some patients by an extensor plantar reflex-positive Babinski sign, characteristics typical of an upper motor neuron disorder. Most of our patients also did not demonstrate significant involvement of the upper limbs, only one had facial muscles involvement and none had elevated CPK levels, as seen in some of the patients in the other studies. With the large size of our cohort and the significant age distribution, our data suggest that these elements are not necessarily characteristic of KY mutations. Similarly, intellectual disability and sensory deficits, found in only few of the patients, may be related to different reasons, genetic or other. The marked phenotypic differences, most prominently the presence of spasticity, delineate a broader range of manifestations, expanding the spectrum of the disease caused by KY mutations from a muscular to a neuromuscular disorder. Similar to our findings, muscle biopsies in both the Israeli–Arab patients and the Kurdish patient showed variability in fiber size, with some internalized nuclei and numerous very small fibers. Variable expression of developmental myosin heavy chain isoforms have been demonstrated in the Kurdish patient, with some small fibers showing abnormal sarcomeres with thickened Z discs and small nemaline rods.14 Electron microscopy of muscle biopsies of the Israeli–Arab patients showed unstructured core targetoid defects with streaming and thickening of the Z discs, as well as an enlarged endoplasmic reticulum, consistent with myofibrillar derangement.15 In line with those findings, the KY-associated phenotype can be classified as a myofibrillar myopathy (MFM7, MIM #617114). It is noteworthy that while NMD was shown in muscle biopsy of one of the c.405C>A patients, both our patients and the one with the c.1071delG variant showed no NMD. Although NMD is not expected in the c.51_52ins(28) variant that creates a frameshift in the first exon,16 NMD could be expected in the two other variants, and was indeed demonstrated in one. Despite irregularity in occurrence of NMD in the different variants, differences in phenotype between patients do not seem related to the presence of KY transcript, as both the c.1071delG and c.405C>A patients have a similar clinical presentation.
Current knowledge of the function of KY protein suggests that it is a transglutaminase-like peptidase, which shows interaction with muscle cytoskeletal proteins such as MYBPC1, TTN, IGFN1 and FLNC, and is part of a Z-band protein complex involved in sarcomere stability.8, 17, 18 Loss of KY function has been shown to affect the cytoskeleton, which presumably leads to the destructive features.17 This is in line with similar HSP syndromes caused by variants in genes related to neuronal cytoskeleton, such as SPAST and CAPN1.19, 20 The mixed myopathic and neurologic features seen in the affected individuals are difficult to explain by loss of kyphoscoliosis peptidase sarcomere stabilizing function alone. Yet, MRI of the head and spinal column done for several patients showed no abnormalities, a finding that is consistent with the normal CNS tissue in mice.9 Nevertheless, KY transcription in CNS tissues (Figure 2c) may imply that the encoded protein may have a yet undiscovered role in neuromotor function, in line with the fact that ky/ky null mutant mice show irregularity of the NMJ.8, 10 Further studies of KY in the nervous system and specifically at the NMJ may uncover new insights as to the role of KY and its malfunction in the interplay between the nervous and muscular systems.
References
Fink JK : Hereditary Spastic Paraplegia Overview. Seattle: University of Washington, 2014, http://www.ncbi.nlm.nih.gov/pubmed/20301682 (accessed 25 November 2016).
de Souza PV, de Rezende Pinto WB, de Rezende Batistella GN, Bortholin T, Oliveira AS : Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 2016; 16: 525–551.
Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Michaela A-G, Stevanin G : Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 2012; 318: 1–18.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P : HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res 2009; 37: W593–W599.
Perez Y, Gradstein L, Flusser H et al: Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet 2014; 22: 703–706.
Silberstein M, Weissbrod O, Otten L et al: A system for exact and approximate genetic linkage analysis of SNP data in large pedigrees. Bioinformatics 2013; 29: 197–205.
Dickinson AG, Meikle VM : Genetic kyphoscoliosis in mice. Lancet (London, England) 1973; 1: 1186.
Blanco G, Coulton GR, Biggin A et al: The kyphoscoliosis (ky) mouse is deficient in hypertrophic responses and is caused by a mutation in a novel muscle-specific protein. Hum Mol Genet 2001; 10: 9–16.
Bridges LR, Coulton GR, Howard G, Moss J, Mason RM : The neuromuscular basis of hereditary kyphoscoliosis in the mouse. Muscle Nerve 1992; 15: 172–179.
Blanco G, Pritchard C, Underhill P et al: Molecular phenotyping of the mouse ky mutant reveals {UCP1} upregulation at the neuromuscular junctions of dystrophic soleus muscle. Neuromuscul Disord 2004; 14: 217–228.
Folker ES, Baylies MK : Nuclear positioning in muscle development and disease. Front Physiol 2013; 4: 363.
Doppler K, Mittelbronn M, Bornemann A : Myogenesis in human denervated muscle biopsies. Muscle Nerve 2008; 37: 79–83.
Altschul SF, Madden TL, Schäffer AA et al: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402.
Carola H-O, Darin N, Engman M et al: A new early-onset neuromuscular disorder associated with kyphoscoliosis peptidase KY deficiency. Eur J Hum Genet 2016; 24: 1771–1777.
Straussberg R, Schottmann G, Sadeh M et al: Kyphoscoliosis peptidase {(KY)} mutation causes a novel congenital myopathy with core targetoid defects. Acta Neuropathol 2016; 132: 475–478.
Pereira FJC, Teixeira A, Kong J et al: Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic Acids Res 2015; 43: 6528–6544.
Beatham J, Romero R, Townsend S, Hacker T, van der Ven P, Blanco G : Filamin C interacts with the muscular dystrophy {KY} protein and is abnormally distributed in mouse {KY} deficient muscle fibres. Hum Mol Genet 2004; 13: 2863–2874.
Baker J, Riley G, Romero RM et al: Identification of a Z-band associated protein complex involving {KY,} {FLNC} and {IGFN1}. Exp Cell Res 2010; 316: 1856–1870.
Trotta N, Orso G, Rossetto MG, Daga A, Broadie K : The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol 2004; 14: 1135–1147.
Gan-Or Z, Bouslam N, Birouk N et al: Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet 2016; 98: 1038–1046.
Acknowledgements
Funding for this research was provided by the Legacy Heritage Biomedical Program of the Israel Science Foundation (grant 1798/16 awarded to OSB and grant 1520/09 awarded to OSB and RB); and by a grant to OSB and scholarship to YY, both by Teva Pharmaceutical Industries Ltd as part of the Israeli National Network of Excellence in Neuroscience (NNE). We thank the patients and their families for their efforts and collaboration in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Yogev, Y., Perez, Y., Noyman, I. et al. Progressive hereditary spastic paraplegia caused by a homozygous KY mutation. Eur J Hum Genet 25, 966–972 (2017). https://doi.org/10.1038/ejhg.2017.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.85
This article is cited by
-
Long-term course of a case with a novel homozygous kyphoscoliosis peptidase variant
Journal of Human Genetics (2024)
-
SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome
European Journal of Human Genetics (2019)
