
Abstract
We report an 8-year-old boy with a complex cerebral malformation, intellectual disability, and complex partial seizures. Whole-exome sequencing revealed a yet unreported de novo variant in the PIK3R2 gene that was recently associated with megalencephaly–polymicrogyria–polydactyly–hydrocephalus (MPPH) syndrome and bilateral perisylvian polymicrogyria (BPP). Our patient showed cerebral abnormalities (megalencephaly, perisylvian polymicrogyria, and mega corpus callosum) that were consistent with these conditions. Imaging also showed right temporal anomalies suggestive of cortical dysplasia. Until now, only three variants (c.1117G>A (p.(G373R)), c.1126A>G (p.(K376E)) and c.1202T>C (p.(L401P))) affecting the SH2 domain of the PIK3R2 protein have been reported in MPPH and BPP syndromes. In contrast to the variants reported so far, the patient described herein exhibits the c.1669G>C (p.(D557H)) variant that affects a highly conserved residue at the interface with the PI3K catalytic subunit α. The phenotypic spectrum associated with variants in this gene and its pathway are likely to continue to expand as more cases are identified.
Similar content being viewed by others

Introduction
Megalencephaly-related (MEG) syndromes are overgrowth disorders presenting with increased brain volume, cortical malformations, developmental vascular anomalies, benign mesenchymal masses, and distal limb malformations.1 Somatic-activating variants in the PI3K–AKT–mTOR pathway have been recently recognized as causative of these overgrowth disorders.2 Postzygotic de novo variants in PIK3CA are responsible for the PIK3CA-related overgrowth spectrum (PROS), including the megalencephaly-capillary malformation syndrome (MCAP) and hemimegalencephaly (HMEG) syndrome.3, 4 De novo germline variants in PIK3R2 and AKT3 genes, which are upstream components of the mTOR pathway, have been found in patients with megalencephaly–polymicrogyria–polydactyly–hydrocephalus syndrome (MPPH).2, 5 More recently, de novo variants in Cyclin D2 (CCND2), a gene downstream of the PI3K–AKT pathway were reported in patients with MPPH,6 whereas germline and somatic variants in PIK3R2 were reported from patients affected by bilateral perisylvian polymicrogyria (BPP).5 Although the phenotypes of these syndromes largely overlap, they can be distinguished based on somatic features because, in contrast to MCAP, MPPH lacks skin vascular malformations, somatic overgrowth, connective tissue dysplasia, and syndactyly.2 Somatic PIK3CA variants are also a common cause of isolated lymphatic malformations in patients without brain involvement.7 The only somatic features reported in MPPH include postaxial polydactyly, found in ~41% of patients, and mild facial dysmorphic features such as prominent forehead, low nasal bridge, and hypertelorism, that are likely secondary to the increased brain volume.1 Additional brain abnormalities reported in these patients include cerebellar tonsillar ectopia (Chiari 1 malformation), whereas increased thickness of corpus callosum was observed in ~7% of cases.1
Here, we report a patient with MPPH syndrome harboring a previously unreported de novo variant in PIK3R2 gene detected by whole-exome sequencing.
Subject and methods
Patient
The reported patient of Italian origin was diagnosed at the Department of Translational Medicine, Section of Pediatrics and the Department of Diagnostic Imaging, Neuroradiology Unit, Federico II University, Naples, Italy. His parents gave written informed consent for this study.
Methods
To uncover genetic variants associated with the abnormalities shown by the patient, we performed whole-exome sequencing of DNA extracted from blood of the proband, both his parents and his unaffected brother (Supplementary Figure S1) as previously described.8, 9 Briefly, exomes were captured using the Agilent SureSelect Human All Exon V4 enrichment kit (Agilent, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq platform. Variants were filtered based on adherence to either an autosomal recessive inheritance pattern or as being de novo in the proband. Further filtering was based on functional candidates among the 33 remaining genes using GeneOntology and OMIM databases. Variants with MAF<0.1 in control populations of European descent and predicted to be deleterious by SIFT10 and/or PolyPhen-211 were prioritized. PCR and Sanger sequencing were used to validate their proper segregation in the family. The model of PIK3R2/PIK3CA interaction was obtained using UNIPROT entry O00459 and Swiss-PdbViewer-DeepView v4.1. All PIK3R2 variants mentioned in the text are described according to transcript NM_005027.3 and protein NP_005018.1. The newly identified PIK3R2 variant was deposited in the Leiden Open Variation Database.
Results
Clinical findings
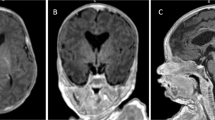
The patient presented with reduced mobility of his left upper arm, developmental delay, and macrocephaly (occipitofrontal circumference (OFC) 48 cm; >98th centile; Supplementary Figure S2A) at 8 months. At 2 years, he showed a left spastic hemiplegia and dysmorphic features (Supplementary Figure S2B), including synophrys, depressed nasal bridge, anteverted nares, pectus excavatum, broad thumb and hallux. A brain MRI, at age 3, revealed hyperplasia of corpus callosum (Figure 1) and asymmetrical bilateral polymicrogyria (PMG; Figure 1a and Supplementary Figure S3A). Reduced myelination of the underlying white matter of the anterior right temporal lobe was suggestive of a focal cortical dysplasia (FCD) type I (Figure 1b). A thick cortical infolding and blurring of a single sulcus of the anterior right insula suggested FCD type IIa; lateral to this lesion, a cyst was identified in the frontal right operculum cortex (Figure 1c and Supplementary Figure S3C). It was unclear how (if) the cystic area was related to the dysplasia. The right thalamus and right cerebral peduncle/brain stem were reduced in size (Supplementary Figure S3B). The left hemisphere had coarse PMG involving perisylvian and suprasylvian cortex (Supplementary Figure S3D). By 4 years of age, the patient developed complex partial seizures characterized by leftward gaze deviation, reduction of muscle tone and loss of consciousness with falls during prolonged epileptic discharges. Antiepileptic therapy with valproic acid, topiramate and clobazam resulted in good seizure control. Interictal-EEG revealed bursts of slow spike/polyspike-and-wave complexes worsening during sleep, especially in right temporal–occipital regions (Supplementary Figure S4).

Brain MRI, localization and modeling of PIK3R2-mutated residue. (a-d) Brain MRI of the patient. (a) T1-weighted axial image: bilateral frontoparietal polymicrogyria and incomplete perisylvian opercularization of the right hemisphere (white arrowhead). (b) T2-weighted axial image: thickness and blurring of the cortex-white matter junction in the right temporal cortex (white arrows) with diminished myelination of the underlying white matter and hypoplasia of right cortico-spinal tract. (c) T1-weighted coronal image shows thick cortical infolding with subtle cortical-white matter blurring in the anterior right insula (white arrow) and a cystic lesion in the right frontal operculum (white arrowhead). (d) T1-weighted mid-sagittal image: increased thickness of corpus callosum. Additional brain MRI images are shown in Supplementary Figure S3. (e) Schematic representation of the PIK3R2 protein domains and positions of the variants reported in literature. Thirty-three cases with c.1117G>A (p.(G373R)) de novo and mosaic variants, one case with a de novo c.1126A>G (p.(K376E)) variant and one case with a p.(L401P) de novo variant2, 5, 12, 13 were reported beside the c.1669G>C (p.(D557H)) patient described here. SH3, Src homology 3 domain; Rho-GAP, Rho GTPase-activating protein domain; SH2, Src homology 2 domain. (f, g) Context of the c.1669G>C (p.(D557H)) variant. The PIK3CA (uniprot P42336) and PIK3R2 (uniprot O00459) are shown in yellow and cyan ribbon diagrams according to the PDB entry 4l2y model, respectively. (f) Detail view of the interaction showing PIK3R2 D557 and PIK3CA N345 forming an H-bond. (g) Groove of ~311 Å3 (in green) with a glycerol molecule present in the pdb entry complex showing PIK3R2 D557 residue in direct contact with its surface. A general view of the interaction between PIK3R2 and PIK3CA and positioning of H557 is shown in Supplementary Figure S6.
Genetic findings
Exome sequencing resulted in five variants that complied with the above filtering (Supplementary Table S1). While we identified in the proband homozygous variants in PSPN and TSEN54, and compound heterozygous variants in CCDC41 that complied with an autosomal recessive inheritance, we also uncovered a de novo variant in the PIK3R2 gene, a gene previously found in patients with MCAP and MPPH.2 The identified variant is a G to C substitution in exon 13 of the PIK3R2 gene (chr19:18,278,049 [hg19]), which modifies a highly conserved aspartic acid (Supplementary Figure S5) into a histidine residue at the amino acid position 557 [NM_005027.3: c.1669G>C; NP_005018.1: p.(D557H)]. Although somatic variants in components of the PI3K–AKT3–mTOR pathway were previously shown to cause hemimegalencephaly,4 the almost 50:50 proportion of G and C nucleotides at position chr19:g.18,278,049 in both high throughput and Sanger sequencing is consistent with a bona fide germline variant. Specifically, we obtained 41 (57%) and 31 (43%) Illumina sequencing reads corresponding to the reference and the alternative (de novo) allele, respectively, whereas the Sanger chromatogram shows equal size peaks (Supplementary Figure S1).
Structural modeling
The PI3K kinase complex is composed of a catalytic moiety, encoded by the PIK3CA, PIK3CB or PIK3CD gene, and one of five different regulatory subunits, such as PIK3R2 (Supplementary Figure S6A). In contrast to the already reported PIK3R2 variants associated with this disorder, (c.1117G>A (p.(G373R)), c.1126A>G (p.(K376E)) and c.1202T>C (p.(L401P)))2, 5, 12, 13 which are positioned within the region encoding the first Src homology 2 (SH2) domain of the protein, the c.1669G>C (p.(D557H)) lies within another interface between the two subunits of the kinase (Figure 1e; Supplementary Figure S6A and B). Both the D557 and the neighboring N561 residues of PIK3R2 generate H-bonds with the side-chain nitrogen atom of the N345 residue of PIK3CA (Figure 1f and Supplementary Figure S6C). It is expected that the c.1669G>C (p.(D557H)) variant would fit nicely in this environment (Supplementary Figure S6D). We hypothesize that the lack of a negative charge does not perturb the interaction as there are no positively charged residues in this portion of the α-catalytic unit. It appears, however, that an ~311 cubic angstroms (Å) groove that could accommodate a small compound is present between the two subunits of the kinase (Figure 1g). Consistent with this hypothesis, in the 4l2y crystal structure, a molecule of glycerol is located at the entry of this groove. The direct contact by the tip of the D557 side chain with the surface of the groove (Figure 1g), as well as the negative charge of this residue could be essential in positioning an adaptor molecule that acts in between the two kinase units in vivo. We hypothesize that a slightly larger and neutral or positively charged histidine variant at this position would result into a smaller groove, as well as altered hydrogen bonds with an adaptor molecule.
Discussion
Although normocephalic patients with PIK3R2 variants were reported,5 our patient shares MEG, bilateral perisylvian PMG, mega corpus callosum, intellectual disability, abnormal muscle tone, spasticity, and epilepsy with the 12 previously described MPPH syndrome patients with PIK3R2 variants.2, 12 Cortical brain malformations were also reported in PROS, particularly in MCAP syndrome with megalencephaly its most consistent feature (OFC>2.5–10 SD).1 Our patient showed a mild asymmetric brain overgrowth, a feature more frequently observed in PROS than MPPH.
The most common pattern of PMG reported in MCAP and MPPH patients is bilateral perisylvian PMG that often extends beyond the perisylvian region.14 Other patterns occur such as focal PMG or PMG involving bilateral frontal lobes.1, 14 Although the extent of PMG involvement is bilateral, many patients show a mildly asymmetric presentation, as observed in our case.5 The proband’s brain MRI revealed PMG with broad cerebral involvement extending to the frontoparietal cortex; in addition a sulcus with cortical-white matter blurring in the anterior right insula and abnormal white matter signal in right anterolateral temporal lobe, and an infolding of thick cortex with cortical blurring in the right frontal operculum are consistent with FCD,15 a malformation not yet described in MPPH. Nevertheless, FCD was observed in PROS, highlighting the overlap between these clinical entities.3 Brain-mosaic-activating variants of PI3K–AKT–mTOR were identified in patients with isolated FCD type II, HMEG, MEG and intractable epilepsy without MRI-identifiable lesions.16, 17 The patient brain MRI also revealed a small area with cyst-like appearance, adjacent to the region of dysplastic cortex in the frontal right operculum that could represent either a small dysembryoplastic neuroepithelial tumor or other cyst, a feature not reported in previous cases. Considering the risk of brain cancer,2 an ongoing surveillance should be considered.
Epilepsy is another distinctive phenotypic characteristic of MPPH syndrome. A wide spectrum of seizures was reported ranging from complex partial seizures to infantile spasms.5 The seizure presentation is predominantly focal, even if no clear recurrent epilepsy pattern is described. In fact, atypical absences, myoclonic jerks, generalized tonic–clonic or complex febrile seizures are reported in these individuals.5, 14 Our patient showed focal seizures characterized by dyscognitive features with a critical onset in the right hemisphere. As described in asymmetrical bilateral PMG, our case had epilepsy laterality with more severe and difficult-to-treat seizures.18
In absence of consistent dysmorphic features in MPPH syndrome, it is worthwhile pinpointing phenotypic characteristics, including synophrys, broad-looking thumbs, large great toes, shared by our patient with two cases recently described.5
Although the PIK3R2 de novo variant identified in the proband does not lie within the SH2 domain, whose variants were previously associated with this disorder, it is the most likely causative variant as it affects a highly conserved residue at the interface with PIK3CA. This variant putatively modifies the size and shape of the groove separating the two kinase subunits.
This case extends the spectrum of MPPH syndrome and highlights the role of the interface domain of PIK3R2.
References
Mirzaa GM, Riviere JB, Dobyns WB : Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet 2013; 163C: 122–130.
Riviere JB, Mirzaa GM, O'Roak BJ et al: De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012; 44: 934–940.
Keppler-Noreuil KM, Rios JJ, Parker VE et al: PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A 2015; 167A: 287–295.
Lee JH, Huynh M, Silhavy JL et al: De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012; 44: 941–945.
Mirzaa GM, Conti V, Timms AE et al: Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol 2015; 14: 1182–1195.
Mirzaa GM, Parry DA, Fry AE et al: De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet 2014; 46: 510–515.
Luks VL, Kamitaki N, Vivero MP et al: Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr 2015; 166: 1048–1054, e1041-1045.
Alfaiz AA, Micale L, Mandriani B et al: TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat 2014; 35: 447–451.
Borck G, Hog F, Dentici ML et al: BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res 2015; 25: 609.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Nakamura K, Kato M, Tohyama J et al: AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin Genet 2014; 85: 396–398.
Tapper WJ, Foulds N, Cross NC et al: Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PLoS ONE 2014; 9: e86940.
Mirzaa GM, Conway RL, Gripp KW et al: Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A 2012; 158A: 269–291.
Blumcke I, Thom M, Aronica E et al: The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011; 52: 158–174.
Jansen LA, Mirzaa GM, Ishak GE et al: PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015; 138: 1613–1628.
Lim JS, Kim WI, Kang HC et al: Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015; 21: 395–400.
Shain C, Ramgopal S, Fallil Z et al: Polymicrogyria-associated epilepsy: a multicenter phenotypic study from the Epilepsy Phenome/Genome Project. Epilepsia 2013; 54: 1368–1375.
Acknowledgements
Authors thank the family for its contribution and the members of the Lausanne Genomic Technologies Facility. AAA is recipient of a scholarship from the Saudi Arabian National Guard Health Affairs. This work was supported by a grant of the Swiss National Science Foundation 31003A_160203 and the Lithuanian-Swiss Cooperation Programme (AR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Note added in proof
During the revision of this manuscript, Mirzaa et al.5 reported PIK3R2 variants in BPP, we modified our manuscript to discuss our results in view of these recently published data to help potential readers getting a complete view of current knowledge.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Terrone, G., Voisin, N., Abdullah Alfaiz, A. et al. De novo PIK3R2 variant causes polymicrogyria, corpus callosum hyperplasia and focal cortical dysplasia. Eur J Hum Genet 24, 1359–1362 (2016). https://doi.org/10.1038/ejhg.2016.7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.7
