
Abstract
Genome-wide association studies (GWAS) in migraine are providing the molecular basis of this heterogeneous disease, but the understanding of its aetiology is still incomplete. Although some biomarkers have currently been accepted for migraine, large amount of studies for identifying new ones is needed. The migraine-associated variant rs12355831:A>G (P=2 × 10−6), described in a GWAS of the International Headache Genetic Consortium, is localized in a non-coding sequence with unknown function. We sought to identify the causal variant and the genetic mechanism involved in the migraine risk. To this end, we integrated data of RNA sequences from the Genetic European Variation in Health and Disease (GEUVADIS) and genotypes from 1000 GENOMES of 344 lymphoblastoid cell lines (LCLs), to determine the expression quantitative trait loci (eQTLs) in the region. We found that the migraine-associated variant belongs to a linkage disequilibrium block associated with the expression of an acyl-coenzyme A synthetase 5 (ACSL5) transcript lacking exon 20 (ACSL5-Δ20). We showed by exon-skipping assay a direct causality of rs2256368-G in the exon 20 skipping of approximately 20 to 40% of ACSL5 RNA molecules. In conclusion, we identified the functional variant (rs2256368:A>G) affecting ACSL5 exon 20 skipping, as a causal factor linked to the migraine-associated rs12355831:A>G, suggesting that the activation of long-chain fatty acids by the spliced ACSL5-Δ20 molecules, a mitochondrial located enzyme, is involved in migraine pathology.
Similar content being viewed by others
Introduction
Migraine is the most frequent type of headache in children. It also affects approximately 14% of adult population, being more frequent in women (20%) than in men (8%). It has a strong genetic basis with heritability estimated of 40–57%.1 The aetiology of migraine is starting to be understood as a result of basic-clinical investigations and genome-wide association studies (GWAS) applied to common form of migraine.2, 3, 4, 5 These investigations aim at getting biomarkers and clinical targets for different types of migraine.6, 7, 8, 9
An example of a biomarker currently accepted for the common migraine with aura subtype is the MTHFR C677T polymorphism, for which genetic testing is currently available and a nutraceutical treatment is known to alleviate symptoms.10 Other classic example includes the neuropeptide calcitonin gene-related peptide (CGRP) involved in migraine, which are significantly associated with response to specific treatments for acute migraine attacks and prophylaxis. CGRP can stimulate the release of pro-inflammatory cytokines, some of which are established to contribute to both the risk of and the outcome from cerebrovascular disease.11
One of the largest migraine GWAS data set currently available3 from the the International Headache Genetics Consortium performed on 29 populations, including a total of 23 285 individuals with migraine and 95 425 controls, identified 142 polymorphisms located in 12 different loci, significantly associated with the phenotype. Some of the loci point to new genes (near AJAP1 on 1p36, near TSPAN2 on 1p13, within FHL5 on 6q16, within c7orf10 on 7p14, and near MMP16 on 8q21) and others confirm previous reports. Three of these loci were identified in disease subgroup analyses. Expression quantitative trait loci (eQTLs) analysis performed on 394 brain tissue specimens revealed 5 additional loci potentially implicated in migraine susceptibility (APOA1BP, TBC1D7, FUT9, STAT6 and ATP5B). Some additional loci showed suggestive evidence for association in the combined data (P<1 × 10−6), but failed to surpass the genome-wide significance threshold. Different functional studies3, 8, 9 suggest the involvement of pathways for glutamatergic neurotransmission, synaptic function, pain sensing, metalloproteinases, and the vasculature, key elements in the anatomical alterations seen in the migraine brain.
The complexity of migraine pathology with features that may have different genetic causes12, 13 complicates the discovery of genuinely-associated genes. In addition, one important reason to fail in assigning causal genes of complex diseases by GWAS is the fact that the associated signals used to be found in non-coding sequences, often containing multiple genes, and the proximity criteria to the top hits, used for selecting putative disease-responsible genes, are not always correct. Some of these genes may actually represent the genetic alteration responsible for the disease association, but for most of them, they merely tag a block of several variants in linkage disequilibrium (LD) in a nearby gene.14, 15
Furthermore, current GWAS top hits of complex conditions explain only a small part of the disease heritability; and therefore, genes identified in this way reflect only a fraction of the pathways conferring genetic disease risk. Rare or family-specific variants have been missed using GWAS approach and could explain the lack of hits to date. As many variants with small effect size are likely to confer disease risk, very large sample sizes, perhaps even hundreds of thousands of individuals are needed to provide sufficient statistical power.15, 16 Finally, positive signals with subGWAS level of significance (P-values>5 × 10−8) constitute another subgroup of variants that may represent truely-associated loci and it is worth to restudy in larger sample sizes or disease subtypes.17
In this study, we analysed the migraine-associated variant rs12355831:A>G, localized at chr10:g.114202526 (hg19), reported in a migraine GWAS performed by the International Headache Genetic Consortium.3 This migraine-associated variant was located in an intronic region of ZDHHC6 gene, with unknown function, and it was studied considering an expression regulatory role. We found a functional variant (rs2256368:A>G) in the LD block tagged by the migraine-associated polymorphism, in the intron 20 of ACSL5 gene (NM_016234.3), and showed it was responsible for the ACSL5 exon 20 skipping. This exon 20 spliced ACSL5 RNA isoform may produce a protein that lacks the peptide corresponding to the sequence of exon 20 (splice variant ACSL5-Δ20). We discuss a hypothetical pathogenic mechanism involving the possible dysfunctional enzymatic activity of the splice variant ACSL5-Δ20 in the frame of the evidence, indicating a disarrangement of the mitochondrial energy metabolism and apoptosis mechanisms in migraine.
Materials and methods
Gene expression and genotype integration analysis
For this study, we used the data from RNA-Seq from European lymphoblastoid cell lines (LCLs) of the GEUVADIS RNA sequencing Project (ftp://ftp.ebi.ac.uk/pub/databases/microarray/data/experiment/GEUV/E-GEUV-1/analysis_results/).18 For the eQTLs determination, we used the genotypes of the same LCLs, obtained from the 1000 Genomes Project19 (phase 1 release v.3) (http://www.1000genomes.org/) as previously reported.20 For Gene Expression Profiling analysis, reads were mapped to the human reference genome (assembly GRCh37.68) as reported.20
Nomenclature and database submission
The nomenclature system for the description of the DNA variant rs2256368:A>G and the corresponding changes in RNA and protein follows the recommendations of the Human Genome Variation Society, with numbering starting at the first position of the translation initiation codon of the GenBank RefSeq NM_016234.3. Thus, rs2256368:A>G was located in chr10:g.114186624:A>G (hg19), in the intron 20 of ACSL5 gene (ACSL5 c.2079+7G>A), whose G allele affects exon 20 skipping (ACSL5 c.2007_2079del), potentially translated into a protein sequence with exon 20 deleted (p.Val671_Val694del). This variant information has been submitted to the LOVD 3.0/shared (http://databases.lovd.nl/shared/view/ACSL5) with the submission ID #60251.
Reverse transcriptase and quantification by qPCR
Total RNA from different LCLs carrying the three rs2256368:A>G genotypes (CODE, AA genotype: GM12006E, GM11994E, GM12043, GM11993D; AG: GM12004D, GM12044E, GM12144D, GM12717L; GG: HG00134, HG00326, HG01048, HG01383) was extracted and processed as previously reported.20 ACSL5 RNA quantifications by relative qPCR were normalized to UBE2D2 mRNA levels, using 2E ΔCt (ΔCt=Ct sample−Ct reference) method as described.21 The primer sequences were designed using Primer3 browser (key: forward-Fw; reverse-Rv; E, exon; 5'-3' direction): UBE2D2 Fw-CAATTCCGAAGAGAATCCACAAGGAATTG and Rv-GTGTTCCAACAGGACCTGCTGAACAC; non-Spliced E20 (using bridge E19–E20 to E21) Fw-CCAAGTTGTAAGGGAAGCCA and Rv-GCTGTCAATTTGGGTCCGAA; Spliced E20 (using bridge E19–E21 to E21) Fw-ACTGTGCCAAAACCAAGTCA and Rv-TGTGCTCATACAGGCTGTCA; and Non-Spl (E19–E21) Fw-CTTCCCTCATTTGCAGCCAA and Rv-GCTCTCCTCGCTTTGCTTTC. Thermocycling conditions for all these amplifications were 1 × (95 °C, 3 min); 30 × (95 °C for 20 s, 59 °C for 20 s, 72 °C for 20 s).
Exon splicing assay
Alternative E20 splicing analysis was performed as previously described.20, 22 Briefly, we carried out a PCR amplification of a genomic DNA fragment (698 bp) containing the E20 of ACSL5 gene (73 bp) and flanking intronic sequences (335 bp at the 5′ and 290 bp at the 3′ of the exon 20) from the LCL GM12004D (rs2256368-AG) using the following oligonucleotides: Fw- 5′-GCAGCCCAAACAGACTGAA and Rv- 5′-TGCTCTGTGAAGAAAGTGAGG. This was performed by Expand High Fidelity PCR System (Roche Diagnostics GmbH, Mannheim, Germany) under the following PCR thermocycling conditions: 1 × (94 °C for 2 min); 30 × (94 °C for 15 s, 59 °C for 30 s, 72 °C for 45 s); 1 × (72 °C for 7 min). The PCR products were purified from 2% agarose-gel electrophoresis with QiaexII gel extraction kit (QIAGEN GmbH, Hilden, Germany), cloned in pCR-2.1 vector (TOPO-TA Cloning Kit, Invitrogen, Carlsbad, CA, USA) and sequenced to identify the clones carrying each allele and to discard potential PCR errors. An EcoRI fragment from each TOPO vector was again purified from agarose-gel electrophoresis and subcloned into the EcoRI site of the pSPL3 minigene plasmid, which was previously cut with EcoRI and purified from agarose gel using QiaexII gel extraction kit (QIAGEN GmbH), and dephosphorylated using FastAP Thermosensitive Alkaline Phosphatase (ThermoFisher Scientific Inc, Leicestershire, UK). Colonies were checked by sequencing for pSPL3 plasmids containing inserts in the right orientation with respect to the promoter of the vector. The pSPL3 plasmids bearing ‘A’ and ‘G’ allele fragments (pA, pG) and plasmid without insert (p) were transfected in HEK cells using JetPrime Transfection reagent (Polyplus Transfection SA, Illkirch-Graffenstaden, France) and harvested 24 h after transfection. After RNA extraction, the cDNA was produced using oligo-dT with SuperScript III First Strand Synthesis Super Mix (Invitrogen) and amplified by PCR using vector-specific primers SD6 (5′-TCT GAG TCA CCT GGA CAA CC-3′) and SA2 (5′-ATC TCA GTG GTA TTT GTG AGC-3′) under the following thermocycling conditions: 1 × (95 °C for 2 min); 30 × (95 °C for 20 s, 58 °C for 30 s and 72 °C for 1 min); 1 × (72 °C for 7 min). The PCR products were sequenced and visualized in 2% agarose-gel electrophoresis.
Statistical calculations
We conducted Spearman's rank correlation analyses with single-nucleotide polymorphisms (SNPs) from the 1000 Genomes Project phase 1 release v3 data set with MAF (minor-frequent allele) >0.05 and the transcript expression levels from GEUVADIS. The setting of an MAF of ⩾0.05, excluding all rare variants to detect eQTLs, was necessary as the transcriptomes used to determine the eQTLs were from 344 subjects (LCLs from 1000 Genomes). MAF <0.05 would have not power enough to obtain eQTLs of rare variants. A P-value of false discovery rate was obtained as previously indicated.10 Quantification by qPCR experiments was performed in triplicates and two to four independent experiments.
Results
Different GWAS variants in the analysed region
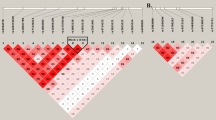
The genomic region analysed in this study (Figure 1) has been associated with different diseases by GWAS, as it is indicated in the UCSC Genome Browser, such as blood lipid levels (rs2255141); migraine (rs12355831); colorectal cancer (rs12241008); and multiple sclerosis brain lesion distribution (rs17267338). As all of these SNPs were located either in intronic or in intergenic non-coding sequences, we designed this study considering their expression regulatory role.

Genomic region in the chromosome 10 associated with different diseases. As obtained from UCSC Genome Browser, the scheme shows from top to bottom: the scale and position, the set of variants (blue) associated with the expression of the ACSL5 transcripts (red), in high LD with the migraine-associated rs12355831:A>G SNP (green), and the Ref-Genes in the region (dark blue).
Linkage disequilibrium in the region and eQTLs
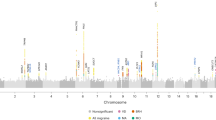
We first searched for SNPs associated with gene expression (eQTLs) by means of processing the RNA-sequencing reads from GEUVADIS Project18 and the genotypes of the same 344 LCLs from 1000 Genomes Project.19 This integration of data sets resulted in the finding of a high LD block (r2>0.8) (Figure 2) of a set of variants, including the migraine-GWAS rs12355831:A>G, but not the other GWAS SNPs in the region, associated with changes in the expression of two ACSL5 transcripts (ENST00000433418, ENST00000356116) from Ensembl that showed alternative splicing of the exon 20 (E20).

Linkage disequilibrium (LD) of the GWAS variants with the best eQTL. LocusZoom plots showing the LD (colour scales) of the GWAS/disease-associated variants, with the best eQTL-associated SNP in the ACSL5 locus, rs2256368:A>G, obtained from 1000 Genomes EUR population (arrows). Only the migraine-associated GWAS variant was in total LD (r2=1) with the rs2256368:A>G SNP. Colour scales represent the LD intervals (r2 values).
Expression association
Among the variants in the LD block, we selected rs2256368:A>G as the best eQTL in total LD (r2=1) with the migraine-associated variant, located at 7 bases downstream of the splicing acceptor site of E20. This position has been described as essential for the interaction with the U1 snRNP component of the spliceosome;23 and therefore, it was considered as a potential candidate that could affect the splicing of the E20.
ACSL5 RNA isoforms quantification was performed as previously described,20 indicated in the figure as FPKM (fragments per kilobase of exon per million fragments mapped), with and without E20, in 344 LCLs versus the rs2256368:A>G genotypes (Figure 3), showing opposite correlations with the genotypes of the best ACSL5 eQTL variant. Thus, the colocalization by LD of the migraine-GWAS variant and the best ACSL5 QTL implied that both signals point to the same causality.

Correlation of the rs2256368:A>G genotypes with the expression levels of two ACSL5 RNA isoforms in 344 LCLs, represented in box plots. Data were obtained by integration of the GEUVADIS RNA-sequencing data and 1000 Genomes genotypes of the same LCLs. Statistical analysis was performed by Spearman's correlation test. Correlation index (rho) and P-values (p) are indicated inside the plots.
Quantification of ACSL5 RNA isoforms
To quantify the expression levels of the two ACSL5 RNA isoforms and to validate the data obtained by integrating the RNA-Seq from GEUVADIS and genotypes from 1000 Genomes, we performed real-time RTqPCR with RNA from 12 LCLs of known genotypes for the variant rs2256368:A>G (Figure 4). Genotype GG was infrequent in 503 subjects from EUR population (F=0.002), getting only four LCLs with rs2256368-GG genotypes in the whole LCL collection. These results were also visualized in polyacrylamide gel electrophoresis after performing end-point classic PCR, using bridge primers for E19–E20 (non-spliced E20) and E21–E19 (spliced E20). As observed in Figure 4, ACSL5 transcripts lacking E20 were expressed about 16- to 31-fold higher in cells bearing GG than AA genotypes, as resulted from qPCR value comparisons of GG versus AA genotypes in Figure 4b. Cells bearing GG genotypes expressed 27–43% of spliced E20 with respect to non-spliced E20 isoforms as calculated from the qPCR values of GG in Figure 4b versus GG in Figure 4a.

Quantification of the ACSL5 RNA isoforms from different LCLs carrying the three rs2256368:A>G genotypes. Classic PCR amplification visualized by acrylamide-gel electrophoresis and relative real-time qPCR values, reflecting the ratio between the expression levels of the ACSL5 transcripts versus the levels of the reference gene, are indicated in the following panels: (a) ACSL5 transcript levels containing E20 (non-spliced E20) by amplification with a bridge primer in E19–E20 to E21; (b) ACSL5 transcript levels lacking E20 (spliced E20) by amplification with a bridge primer in E19–E21 to E21; (c) ACSL5 transcripts with and without E20 by amplification with primers E19–E21.
Direct causality of rs2256368:A>G variant in E20 skipping
To confirm that the rs2256368:A>G is the causal variant of the E20 splicing observed in the ACSL5 transcript profile, we used a strategy by cloning the E20 and its flanking intronic sequences, carrying as unique difference the two rs2256368:A>G alleles, into the pSPL3 plasmid (Figure 5). After transfection in HEK cells, RNA purification, RT-PCR amplification, analysis of the RNA products by agarose-gel electrophoresis and sequencing, we determined that the E20-G allele was spliced in about 40% of the molecules and <5% of the molecules in the E20-A allele. These splicing percentages varied in different experiments in which the length of the flanking intronic sequences of E20 was changed (not shown). Therefore, these data were in agreement with the previous results of this study, confirming that the rs2256368-G allele was associated with splicing alterations resulted in the expression of ACSL5 RNA isoforms lacking E20 (ACSL5-Δ20).

E20 skipping analysed by alternative splicing construct assay. (a) The scheme represents the fragment (introns: horizontal lines; exons: boxes; fragment sizes in bp) obtained by PCR amplification of the ACSL5 exon 20 (grey box) with intron flanking sequences and the position of the polymorphism rs2256368:A>G (bold), cloned in the multi-cloning site position (m.c.s.) of the pSPL3 vector. (b) Agarose-gel electrophoresis of RT-PCR amplification of the RNA purified from HEK cells transfected with the constructs carrying the rs2256368-G (pG) or A alleles (pA) and control plasmid (p). Lane m stands for the DNA size markers.
Discussion
In this work, we have shown the SNP rs2256368:A>G, in total LD (r2=1) with migraine GWAS-associated rs12355831:A>G3, as the causal genetic determinant responsible for the expression of ACSL5 transcript variant lacking E20 (ACSL5-Δ20). Although activity of eQTLs can be cell type and stimulus dependent,20, 21 the ACSL5 enzyme (EC 6.2.1.3) is expressed in all tissues and cells at different levels, some times under specific cell stimulation.21, 24 So it is very likely that the studied eQTL is also operating in brain tissue, where migraine disorder takes place.
This enzyme is localized in the mitochondrial outer membrane and microsomes25 where it catalyses the activation of long-chain fatty acids by thioesterification with coenzyme A (CoA). ACSL5 is thus having an important role in supplying exogenous fatty acids into the mitochondria (mt) for oxidative reactions, increasing ceramide synthesis, acylation of proteins, and affecting the mitochondrial membrane potential.26, 27, 28 Furthermore, ACSL5 has been involved in the regulation of cell growth, pro-apoptotic sensing of enterocytes,26 and mitogen-activated lymphocytes.24 Human ACSL5 is shown to be experimentally inhibited by triacsin C29 and may be the target of drug actions as shown in different experimental systems.29, 30, 31
Given the limited knowledge on the functional activity of the ACSL5Δ20 enzyme, it is difficult to envisage the pathogenic relevance in migraine of the risk rs2256368-GG carriers, ‘high producers’ of ACSL5-Δ20. As described by Gassler et al.,26 in contrast with the splice variant ACSL5-Δ20, recombinant and purified full-length enzyme is active at high alkaline pH. Therefore, the genetic factor associated with pathogenesis of the migraine may be related with some type of dysfunction of the protein lacking E20 peptide. On the other hand, the fact that ACSL5 is the only member of the ACSL family (including ACSL1, 3, 4, 5 and 6) located in the mt, indicates that the enzymatic activity affected in the splice variant ACSL5Δ20 is relevant to migraine pathology only in this suborganelle.
The hypothesis of the mitochondrial component in migraine neurobiology has been supported in several studies,32, 33, 34, 35, 36, 37 suggesting that an impairment of the mitochondrial oxidative metabolism, which ultimately causes energy failure in neurons and astrocytes, triggers migraine mechanisms. Furthermore, different types of assays performed in migraine sufferers have shown a decreased activity of the respiratory chain enzymes33 and a correlation between the extent of the energy disturbance (low ATP levels), and the clinical phenotype severity.37 Our data suggest that the acyl-CoAs produced by ACSL5 enzyme or the downstream metabolism of these activated fatty acids (for beta-oxidation, mitochondrial membrane integrity, and protein acylation) could be altered by the presence of the splice variant ACSL5Δ20, as carriers of the rs2256368-GG genotype produced about 40% of ACSL5 molecules lacking E20.
Several compounds such as riboflavin/vitamin B2, CoQ10, magnesium, niacin, carnitine, topiramate, and thioctic acid/lipoic acid, that have a positive effect on mitochondrial metabolism, have been shown to prevent recurrent headaches.34, 35 This evidence is in agreement with the hypothesis of dysfunctional energy metabolism, in which ACSL5 is implicated by supplying activated fatty acids for beta-oxidation or acylation of other mitochondrial proteins and ceramides. Specific mitochondrial DNA mutations have not been detected in patients with migraine;33 thus, this ACSL5 rs2256368:A>G would be an SNP of a nuclear gene encoding for a mitochondrial protein.
Another hypothesis of how the splice variant ACSL5-Δ20 could be involved in a mitochondrial dysfunction might be suggested from the acyl-CoA synthetase-1 study in the malaria parasite Plasmodium falciparum (PfACS1).38 This protein physically interacts through its carboxyl-terminal domain with the erythrocyte ankyrin, as part of its role in capturing serum fatty acids for activation and posterior utilization by the intracellular parasites. This ankyrin-interacting domain in PfACS1 is likely the equivalent region of the human ACSL5-E20. It is tempting to speculate that the lack of E20 could affect the process of protein–protein interactions of ACSL5 with other enzymes involved in the processing of the acyl-CoAs in the mt and/or microsomes. As a matter of fact, ACSL5 mediates antiproliferative activities via Wnt2B palmitoylation in the mt with diminished Wnt activity.39
In support of the hypothesis, ACSL5 interacts with ceramide synthases (CerS) and diacylglycerol acyltransferase 2 (DGAT2) to form a ACSL5–CerS–DGAT2 complex,40 in which ACSL5 provides activated long-chain fatty acids, CerS generates de novo synthesis of ceramides and the DGAT2 catalyses the transference of the fatty acids to ceramides, to finally produce acyl-ceramides. If channelling of novo ceramides to acyl-ceramides is inhibited, then an increase in ceramide-mediated apoptosis is produced.41 Spliced ACSL5-Δ20 could be unable to form the tri-molecular complex, and as a result, decreasing the acyl-ceramides produced. A recent biomarker study42 has shown that ceramides were decreased in the blood of women with episodic migraine as compared with those women without any headache disorders. If confirmed, the link between the SNP rs2256368:A>G and a low production of acyl-ceramides may take place as a consequence of a dysfunctional spliced ACSL5-Δ20, supporting its implication in migraine pathology.
Gene-based analysis of the migraine GWAS data, using detailed spatial gene expression data in the normal human brain,43 have shown moderate enrichment of migraine-associated genes in modules involved in cortical neurotransmission, mitochondrial and oligodendrocyte function that provide further evidence that these mechanisms have a causal role in migraine, and again supporting the molecular activity reported for ACSL5 enzyme in apoptosis and cell differentiation.
In summary, this study contributes to the identification of a functional SNP (rs2256368:A>G) in total LD with the migraine GWAS-associated variant (rs12355831:A>G). Carriers of rs2256368-GG produce a measurable molecular phenotype affected by E20 skipping in the transcripts of ACSL5 gene. Hypothetically, a spliced ACSL5-Δ20 enzyme, lacking amino-acid sequence of E20 (p.Val671_Val694del), could be unable of interacting with CerS and DAGT2 complex to form acyl-ceramides. These lipids are involved in early apoptosis induced from mt and are decreased in blood of women with sporadic migraine. This mechanism is congruent with the activity that ACSL5 performs in the energy metabolism, synthesis of acyl-ceramides and mitochondrial apoptosis, suggesting that the spliced ACSL5-Δ20 is implicated in the pathogenesis of migraine.
References
Mulder EJ, Van Baal C, Gaist D et al: Genetic and environmental influences on migraine: a twin study across six countries. Twin Res 2003; 6: 422–431.
Anttila V, Stefansson H, Kallela M et al: Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010; 42: 869–873.
Anttila V, Winsvold BS, Gormley P et al: Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet 2013; 45: 912–917.
Chasman DI, Schurks M, Anttila V et al: Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet 2011; 43: 695–698.
Freilinger T, Anttila V, de Vries B et al: Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet 2012; 44: 777–782.
Durham P, Papapetropoulos S : Biomarkers associated with migraine and their potential role in migraine management. Headache 2013; 53: 1262–1277.
Tan MS, Jiang T, Tan L, Yu JT : Genome-wide association studies in neurology. Ann Transl Med 2014; 2: 124.
Tolner EA, Houben T, Terwindt GM et al: From migraine genes to mechanisms. Pain 2015; 156 (Suppl 1): S64–S74.
Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AM : Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol 2015; 14: 65–80.
De Vries B1, Haan J, Frants RR, Van den Maagdenberg AM, Ferrari MD : Genetic biomarkers for migraine. Headache 2006; 46: 1059–1068.
Denes A, Pinteaux E, Rothwell NJ et al: Interleukin-1 and stroke: Biomarker, harbinger of damage, and thera- peutic target. Cerebrovasc Dis 2011; 32: 517–527.
Chasman DI, Anttila V, Buring JE et al: Selectivity in genetic association with sub-classified migraine in women. PLoS Genet 2014; 10: e1004366.
Ligthart L, Hottenga JJ, Lewis CM et al: Genetic risk score analysis indicates migraine with and without comorbid depression are genetically different disorders. Hum Genet 2014; 133: 173–186.
Khankhanian P, Gourraud PA, Lizee A, Goodin DS : Haplotype-based approach to known MS-associated regions increases the amount of explained risk. J Med Genet 2015; 52: 587–594.
Eising E, de Vries B, Ferrari MD, Terwindt GM, van den Maagdenberg AM : Pearls and pitfalls in genetic studies of migraine. Cephalalgia 2013; 33: 614–625.
Potenciano V, Abad-Grau MM, Alcina A, Matesanz F : A comparison of genomic profiles of complex diseases under different models. BMC Med Genomics 2016; 9: 3.
Lill CM, Luessi F, Alcina et al: Genome-wide significant association with seven novel multiple sclerosis risk loci. J Med Genet 2015; 52: 848–855.
Lappalainen T, Sammeth M, Friedlander MR et al: Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013; 501: 506–511.
1000 Genomes Project Consortium, Abecasis GR, Auton et al: An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Matesanz F, Potenciano V, Fedetz M et al: A functional variant that affects exon-skipping and protein expression of SP140 as genetic mechanism predisposing to multiple sclerosis. Hum Mol Genet 2015; 24: 5619–5627.
Karaky M, Alcina A, Fedetz M et al: The multiple sclerosis-associated regulatory variant rs10877013 affects expression of CYP27B1 and VDR under inflammatory or vitamin D stimuli. Mult Scler 2015, pii: 1352458515610208.
Desviat LR, Perez B, Ugarte M : Minigenes to confirm exon skipping mutations. In exon skipping: methods and protocols. Methods Mol Biol 2012; 867: 37–47.
Ward AJ, Cooper TA : The pathobiology of splicing. J Pathol 2010; 220: 152–163.
Catalá-Rabasa A, Ndagire D, Sabio JM, Fedetz M, Matesanz F, Alcina A : High ACSL5 transcript levels associate with systemic lupus erythematosus and apoptosis in Jurkat T lymphocytes and peripheral blood cells. PLoS One 2011; 6: e28591.
Mashek DG, McKenzie MA, Van Horn CG, Coleman RA : Rat long chain acyl-CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle-RH7777 cells. J Biol Chem 2006; 281: 945–950.
Gassler N, Roth W, Funke B et al: Regulation of enterocyte apoptosis by acyl-CoA synthetase 5 splicing. Gastroenterology 2007; 133: 587–598.
Kagan VE, Borisenko GG, Tyurina YY et al: Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med 2004; 37: 1963–1985.
Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K : Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol 2015; 21: 3492–3498.
Kaemmerer E, Peuscher A, Reinartz et al: Human intestinal acyl-CoA synthetase 5 is sensitive to the inhibitor triacsin C. World J Gastroenterol 2011; 17: 4883–4889.
Kim JH, Lewin TM, Coleman RA : Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem 2001; 276: 24667–24673.
Mashima T, Oh-hara T, Sato S et al: p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. J Natl Cancer Inst 2005; 97: 765–777.
Sparaco M, Feleppa M, Lipton RB, Rapoport AM, Bigal ME : Mitochondrial dysfunction and migraine: evidence and hypotheses. Cephalalgia 2006; 26: 361–372.
D'Andrea G, Leon A : Pathogenesis of migraine: from neurotransmitters to neuromodulators and beyond. Neurol Sci 2010; 31 (Suppl 1): S1–S7.
Markley HG : CoEnzyme Q10 and riboflavin: the mitochondrial connection. Headache 2012; 52 (Suppl 2): 81–87.
Yorns WR Jr, Hardison HH : Mitochondrial dysfunction in migraine. Semin Pediatr Neurol 2013; 20: 188–193.
Colombo B, Saraceno L, Comi G : Riboflavin and migraine: the bridge over troubled mitochondria. Neurol Sci 2014; 35 (Suppl 1): 141–144.
Reyngoudt H, Achten E, Paemeleire K : Magnetic resonance spectroscopy in migraine: what have we learned so far? Cephalalgia 2012; 32: 845–859.
Téllez M, Matesanz F, Alcina A : The C-terminal domain of the Plasmodium falciparum acyl-CoA synthetases PfACS1 and PfACS3 functions as ligand for ankyrin. Mol Biochem Parasitol 2003; 129: 191–198.
Klaus C, Schneider U, Hedberg C et al: Modulating effects of acyl-CoA synthetase 5-derived mitochondrial Wnt2B palmitoylation on intestinal Wnt activity. World J Gastroenterol 2014; 20: 14855–14864.
Senkal C, Hannun Y, Obeid L : Interaction of ceramide synthase with long chain fatty acyl-CoA synthase 5 channels de novo ceramide to acylceramide generation by diacylglycerol acyltransferase 2 on lipid droplets. FASEB J 2015; 29: 1.
Siskind LJ : Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr 2005; 37: 143–153.
Peterlin BL, Mielke MM, Dickens AM et al: Interictal, circulating sphingolipids in women with episodic migraine: a case-control study. Neurology 2015; 85: 1214–1223.
Eising E, Huisman SM, Mahfouz et al: Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: a GWAS-based study using the Allen Human Brain Atlas. Hum Genet 2016; 135: 425–439.
Acknowledgements
We thank the GEUVADIS Consortium for the RNA-Seq data and 1000 Genomes consortium for accessibility that permitted the analysis of our data. We thank Dr LR Desviat for kindly provide us the pSPL3 plasmid and suggestions. This work was supported by Fondo de Investigación Sanitaria (FIS)-Instituto de Salud Carlos III(ISCIII)-Fondos Europeos de Desarrollo Regional (FEDER), Unión Europea (grant numbers P12/00555 and PI13/01527) and Junta de Andalucía (JA)- Fondos Europeos de Desarrollo Regional (FEDER) (grant number CTS2704).
Author contributions
FM, MF and CB performed most of the genotyping and expression analysis. VP and FM performed bioinformatics and statistical analysis. MK, AC-R and RB-M performed cell growth and extraction of RNA. FM and AA planning and overseeing the study and writing the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Matesanz, F., Fedetz, M., Barrionuevo, C. et al. A splice variant in the ACSL5 gene relates migraine with fatty acid activation in mitochondria. Eur J Hum Genet 24, 1572–1577 (2016). https://doi.org/10.1038/ejhg.2016.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.54
This article is cited by
-
Role of Omics in Migraine Research and Management: A Narrative Review
Molecular Neurobiology (2022)
-
Changes in the gene expression profile during spontaneous migraine attacks
Scientific Reports (2021)
-
Splice-site variant in ACSL5: a marker promoting opposing effect on cell viability and protein expression
European Journal of Human Genetics (2019)
-
The role of dihydrosphingolipids in disease
Cellular and Molecular Life Sciences (2019)
-
Genetics of hearing loss in the Arab population of Northern Israel
European Journal of Human Genetics (2018)
