
Abstract
Tooth development is controlled by the same processes that regulate formation of other ectodermal structures. Mutations in the genes underlying these processes may cause ectodermal dysplasia, including severe absence of primary or permanent teeth. Four consanguineous Palestinian families presented with oligodontia and hair and skin features of ectodermal dysplasia. Appearance of ectodermal dysplasia was consistent with autosomal recessive inheritance. Exome sequencing followed by genotyping of 56 informative relatives in the 4 families suggests that the phenotype is due to homozygosity for KREMEN1 p.F209S (c.626 T>C) on chromosome 22 at g.29,521,399 (hg19). The variant occurs in the highly conserved extracellular WSC domain of KREMEN1, which is known to be a high affinity receptor of Dickkopf-1, a component of the Dickkopf–Kremen–LRP6 complex, and a potent regulator of Wnt signaling. The Wnt signaling pathway is critical to development of ectodermal structures. Mutations in WNT10A, LRP6, EDA, and other genes in this pathway lead to tooth agenesis with or without other ectodermal anomalies. Our results implicate KREMEN1 for the first time in a human disorder and provide additional details on the role of the Wnt signaling in ectodermal and dental development.
Similar content being viewed by others

INTRODUCTION
Oligodontia (MIM 167416), or severe tooth agenesis, is a rare developmental anomaly characterized by the absence of six or more teeth.1 Oligodontia appears in approximately 1 in 1000 persons and may be isolated or syndromic. Genes responsible for non-syndromic oligodontia, which is generally dominantly inherited, include MSX1 (MIM 142983), PAX9 (MIM 167416), WNT10A (MIM 606268), EDA1 (MIM 300451), and LRP6.2 In contrast, syndromic oligodontia is more often recessively inherited and appears in the context of multiple congenital malformations, reflecting shared developmental pathways of teeth with other orofacial and ectodermal structures.3 Syndromic oligodontia is often a feature of ectodermal dysplasias, which involve anomalies of hair, nails, and sweat glands, in addition to teeth.4 In particular, hypohidrotic ectodermal dysplasias are characterized by reduction in sweat production, abnormal teeth, sparse hair, and thin and dry skin and may be caused by pathogenic variants in WNT10A, EDA1, EDAR (MIM 604095), or EDARADD (MIM 606603). Syndromic oligodontia appears as a feature of multiple forms of ectodermal dysplasias, for many of which the responsible genes have been identified.5
Even given this extensive body of knowledge, new forms of oligodontia continue to appear in the course of dental practice. The present study was motivated by the appearance of severe oligodontia, accompanied by anomalies of hair and skin, first in eight members of two extended kindreds, then in two additional families from the same neighborhood, during the practice of dentistry by one of us (YAI) in Hebron, Palestine. When Sanger sequencing of the most frequently mutated oligodontia genes in the affected individuals revealed normal sequences, we decided to undertake a genome-wide search for a new gene for familial syndromic oligodontia.
MATERIALS AND METHODS
Subjects and clinical examinations
Participants were referred for dental care to the Dr Nidal Maraqa Dental Center, Hebron, Palestine. Inclusion criteria for cases were the presence in oneself or in at least two relatives of syndromic oligodontia with ectodermal dysplasia, defined as the absence of at least six primary and/or permanent teeth, with accompanying hair and skin anomalies. After obtaining written informed consent from adults and from parents of minor children, all participants were evaluated by clinical oral examination. Radiographic examination was carried out for two subjects. Peripheral blood was obtained for extraction of genomic DNA. Controls were 239 Palestinian adults without oligodontia, 39 from the same neighborhood as the study subjects and 200 from other areas of the West Bank. Non-syndromic hypodontia (ie, absence of one to five teeth with no syndromic features) was not excluded as a criterion for participation as a control. The study was approved by the institutional review boards of Bethlehem University, Bethlehem, Palestine, and the University of Washington, Seattle, USA.
Genomics
Genomic DNA extracted from peripheral blood was Sanger sequenced for coding and flanking sequences of MSX1, PAX9, and AXIN2. Coding exons of NFKBIA, a candidate gene identified by homozygosity mapping, were also Sanger sequenced. Following exome sequencing, genotyping of candidate variants was also carried out by Sanger sequencing. PCR products were Sanger sequenced on an ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Primer sequences for Sanger sequencing are listed in Supplementary Table S1. Homozygosity mapping was carried out using the AffyMetrix 250 K Nsp SNP microarray as previously described.6 Exome sequencing was carried out on an Illumina HiSeq2500 (Illumina, Inc., San Diego, CA, USA), as previously described,5, 7 with updated details in Supplementary Methods. Consequences of DNA changes at the RNA level were confirmed by Sanger sequencing of cDNA generated from RNA extracted from white cells of participants. Data were submitted to the Leiden Open Variation Database (LOVD) at www.LOVD.nl/KREMEN1 (submission ID 24090; individual IDs 56411, 56428, 56429, and 56430).
RESULTS
Clinical features
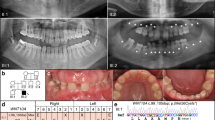
The first patients in the study were two children from the same small town near Hebron, who presented for dental care with oligodontia and other ectodermal features. Evaluation of the children and their immediate relatives suggested a phenotype consistent with autosomal recessive ectodermal dysplasia. In response to inquiries to the community, additional relatives and families presented for care. Ultimately 56 relatives from 4 families were evaluated. Clinical findings for the subjects are summarized in Table 1 and Figure 1, Supplementary Figure S1; the original patients are SO1-IV-2 and SO2-III-6.

Facial and intraoral features of ectodermal dysplasia of the probands of four families. Facial features include abnormal hair distribution of the scalp, low hairline with forehead fuzziness, broad and low nose bridge, columella extending with age, thick lips, slight ocular hypertelorism, and downward slanting of the palpebral fissures. Unaffected relative in these families did not have these features. All affected family member had severe oligodontia of primary and permanent teeth with particular loss of the upper lateral incisors and lower anterior teeth, accompanied by large gaps and reduction of the alveolar bone height.
Affected individuals displayed abnormal hair distribution and texture, with brittle and wiry scalp hair that they described as ‘oily’, and light fuzzy hair on the face and forehead. Eyelashes and eyebrows were present but thin. Facial skin appeared soft, glossy, and thin. Additional facial features present in some but not in all affected individuals included protruded lips, depressed nasal bridge, a broad nose associated with ocular hypertelorism (most pronounced in SO4-II-9 and SO2-III-6), and downward slanting of the palpebral fissures (Supplementary Figure S1). Subjects SO1-IV-1 and SO1-IV-2 have particularly slender limbs and fingers, unlike other members of family SO1. These two siblings were also described as having increased sweating. Subject SO1-IV-4 was described as having dry skin that required frequent ointment application. None of the unaffected relatives in any of the four families displayed any of the features described above.
All affected individuals in the four families displayed severe absence of teeth. Clinical examinations, along with radiographic examinations when available, revealed that the number of missing permanent teeth in these individuals ranged from 8 to 20 (Table 1). The most commonly missing teeth were the upper primary and permanent lateral incisors and the lower primary and permanent anterior teeth. Tooth agenesis was often accompanied by alveolar ridge deficiency and by increased palatal depth. Individual SO4-III-2 displayed the most severe oligodontia with complete agenesis of primary dentition.
In general, affected individuals were born to unaffected parents (Figure 2a). However, in family SO2, the mother (II-8) of three affected children showed isolated oligodontia, with absence of six teeth but no anomalies of the skin or hair (Table 1). Furthermore, in families SO1 and SO2 (but not in families SO3 or SO4), multiple relatives showed isolated hypodontia; that is, loss of one to five teeth with no skin or hair anomalies. In families SO1 and SO2, isolated tooth agenesis differed from syndromic tooth agenesis both in severity and in pattern. The number of missing teeth was significantly greater for family members with syndromic features versus absence of syndromic features (Mann–Whitney test Z=3.79, P=0.0002). The pattern of tooth absence also differed for family members with isolated versus syndromic agenesis. For example, five siblings III-24 through III-28 in family SO1 each lacked one to five permanent teeth, most consistently the second premolars, and six relatives in family SO2 each lacked two to six teeth, most consistently the permanent lower central incisors, followed by the upper lateral incisors (Table 1). Among the subjects with syndromic oligodontia, the most commonly absent teeth were upper lateral incisors and lower anterior teeth, as has been reported for other forms of ectodermal dysplasia.8 Individuals with ectodermal dysplasia showed similar features of hair and skin regardless of the dental status of their parents. Given the multiple causes, both genetic and non-genetic, for isolated tooth agenesis,9 we decided to focus gene discovery on the syndromic phenotype of ectodermal dysplasia including oligodontia.

Ectodermal dysplasia and KREMEN1 p.F209S in four families. (a) Co-segregation of homozygosity for KREMEN1 p.F209S with ectodermal dysplasia in Palestinian families SO1, SO2, SO3, and SO4. Pedigrees indicate individuals with ectodermal dysplasia in black symbols, with the ages at exam (in years) for all individuals indicated under their symbols. Probands are indicated by arrows. Genotypes for KREMEN1 p.F209S are N for the reference allele phenylalanine and V for the variant allele serine at residue 209. (b) Sequence chromatograms of KREMEN1 c.626 T>C (NM_032045.4) for SO1-IV-2 (homozygous for the variant allele), for SO1-III-8 (heterozygous), and for SO1-III-1 (homozygous for the reference allele). (c) Diagram of the human KREMEN1 protein illustrating the functional extracellular Kringle, WSC, and CUB domains; the transmembrane TM domain, and the intracellular IC domain. The position of KREMEN1 p.F209S is indicated with an arrow. Aligned sequences of KREMEN1 orthologs surrounding F209 in the human protein are illustrated under the diagram.
Gene discovery
Regions of homozygosity greater than 2 MB shared by affected siblings SO1-IV-1 and SO1-IV-2 were identified on chromosomes 1p, 5q, 6p, 6q, 10q, 13q, 14q, and 22q (Supplementary Table S2). The longest region of shared homozygosity, 42.4 MB on chromosome 14, harbors at least 435 genes, including NFKBIA, which is responsible for an autosomal dominant anhidrotic ectodermal dysplasia with T-cell immunodeficiency.10 However, Sanger sequencing of genomic DNA from SO1-IV-1 for all coding exons of NFKBIA revealed normal sequence.
Whole-exome sequencing was carried out for affected siblings SO1-IV-1 and SO1-IV-2 and their unaffected parents SO1-III-8 and SO1-III-9. Variants were filtered to identify those that were homozygous in both affected siblings and heterozygous in their parents, and were rare and predicted to be damaging, as described in Supplementary Methods. Three variants met these criteria (Supplementary Table S3). Genotyping these variants in additional affected family members SO1-IV-4, SO1-III-29, and SO1-III-36, who had not been evaluated by exome sequencing, revealed only KREMEN1 c.626 T>C, on chromosome 22 at g.29,521,399 T>C, to be homozygous in all affected relatives in family SO1. This site lies in a 17.4-MB region of homozygosity shared by SO1-IV-1 and SO1-IV-2 (Supplementary Table S2). On the basis of Sanger sequencing of patient-derived cDNA, this change is predicted to lead to substitution of serine for phenylalanine at residue 209. The variant was then genotyped in all participants from family SO1, revealing consistent co-segregation with ectodermal dysplasia under a recessive model (Figures 2a and b). KREMEN1 p.F209S (c.626 T>C) on chromosome 22 at g.29,521,399 T>C has a PolyPhen score of 1.0 and a GERP score of 5.7, has not been reported in any public database and alters a completely conserved residue in the highly conserved WSC domain of KREMEN1 (Figure 2c).
KREMEN1 p.F209 is located at the C-terminal end of the WSC domain. WSC domains are found in a wide variety of proteins from fungi to mammals and are thought to bind carbohydrates.11 In virtually all WSC domains, an aromatic phenylalanine or tyrosine is conserved at the position analogous to KREMEN1 p.F209, with an adjacent hydrophobic valine, leucine, or isoleucine (Supplementary Figure S4). Conservation of this motif across both proteins and species suggests that it is important for the function of the WSC domain. The substitution of a non-aromatic serine for a larger, aromatic phenylalanine or tyrosine at this residue is a highly non-conservative change.
To further evaluate KREMEN1 p.F209S as a candidate for a causal allele for ectodermal dysplasia, we genotyped the variant in 39 unrelated adults from the same neighborhood as family SO1, then used the estimate of the frequency of the variant allele in this local population to estimate the likelihood of co-incidental co-segregation of KREMEN1 p.F209S with ectodermal dysplasia in the remaining families (SO2, SO3, SO4), who also live nearby. Dental examination of the neighborhood controls revealed that 6 of the 39 controls had isolated hypodontia, but these controls were not excluded. Of the 39 controls, 4 were heterozygous for KREMEN1 p.F209S, yielding an allele frequency of 4/78=0.05128. (Among the controls, heterozygosity for KREMEN1 p.F209S was not associated with isolated hypodontia, which was observed in 0/4 controls heterozygous for KREMEN1 p.F209S and in 6/35 controls homozygous for the reference sequence.) Of 200 Palestinian controls from other areas of the West Bank, all were homozygous for the reference allele of KREMEN1 p.F209S.
Next, all participants in families SO2, SO3, and SO4 were genotyped for KREMEN1 p.F209S, revealing perfect co-segregation of the allele with ectodermal dysplasia under a recessive model (Figure 2a). Given an allele frequency of 0.05128 in the local population of these families, the probability of this degree of co-segregation occurring by chance is approximately 2.48 × 10(−15).
DISCUSSION
Three lines of evidence support the interpretation that homozygosity for KREMEN1 p.F209S is responsible for the phenotype of ectodermal dysplasia including oligodontia in these four families. First, KREMEN1 p.F209S is the only rare, putatively damaging, coding sequence variant that is homozygous in all affected family members and not homozygous in family members without this phenotype; P=2.48 × 10(−15) for co-segregation of the variant with the phenotype by chance in the four families. Exome sequencing and pedigree analysis do not preclude the possibility that a variant in complete disequilibrium with KREMEN1 p.F209S, but not detectable by exome sequencing, could be responsible for the phenotype. However, conservation provides strong support for this missense being the critical variant: KREMEN genes exist only in vertebrates,12 and all 100 sequenced vertebrates have phenylalanine at this site (http://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg38&g=cons100way). Finally, as will be explored below, the biology of KREMEN1 strongly supports its involvement in the development of dental and other ectodermal tissues.
KREMEN1 (MIM 609898) encodes a kringle domain-containing transmembrane protein that acts as a receptor for the highly specialized secreted protein Dickkopf-1.11, 13 Dickkopf-1 was first characterized as a potent head inducer in Xenopus embryos, where it was shown to bind LRP6 and strongly inhibit Wnt signaling,14, 15, 16 Kremen-1 and its homolog Kremen-2 bind Dickkopf-1 with high affinity, creating a Dickkopf–Kremen–LRP6 ligand-receptor complex. The interaction of these proteins is physiologically critical to Wnt signaling.17 Although the Wnt signaling pathway is highly conserved throughout evolution, Dickkopf and Kremen proteins are only found in vertebrates, suggesting that the Dickkopf–Kremen–LRP6 complex might be an evolutionarily young regulatory unit that regulates Wnt signaling in vertebrates in coordination with the evolutionarily much older Wnt-Frizzled-LRP6 complex. The observations that Dickkopf-1 interacts with the Kremen extracellular domain, and that the Dickkopf–Kremen–LRP6 ternary complex undergoes rapid endocytosis, suggested that Kremen triggers the internalization and removal of LRP6 from cell surface, inhibiting Wnt signaling.11 In contrast, in the absence of Dickkopf proteins, Kremen can increase Wnt signaling by binding to LRP6 and promoting its cell-surface localization.18 It is proposed that this context-dependent activity of Kremen could potentiate the effect of a shallow gradient of Dickkopf protein to establish robust borders between cell types during development.19 The complex including Dickkopf-1, Kremen, and LRP6 is now recognized as one of the major ligand-receptor complexes that control Wnt signaling in vertebrates.20
Properly regulated Wnt signaling is critical for tooth development, which requires a series of reciprocal signaling interactions between epithelial and neural crest-derived mesenchymal tissues. Wnt signaling is required in both epithelium and mesenchyme. Epithelium-specific inactivation of β-catenin or epithelial expression of Dickkopf-1 causes arrest of tooth development at the early bud stage.21, 22 Mesenchymal-specific elimination of β-catenin causes arrest of tooth development at the bud stage, indicating that Wnt signaling has a role in the transition from the bud to cap stage of developing dental mesenchyme.23 Given the known involvement of WNT10A and LRP6 in tooth development and of pathogenic variants in these genes in oligodontia,2, 24 it is not surprising that mutant KREMEN1 also has a role.
We speculate that the substitution of serine for phenylalanine in the KREMEN1 WSC extracellular domain interferes with KREMEN1 regulation of Wnt signaling, either as a positive or as a negative regulator, or possibly both at different stages of development. In Xenopus, KREMEN2 has been shown to have Dickkopf-independent activity as an activator of Wnt signaling that is required for the formation of neural crest tissue.14 This observation is consistent with the finding that oligodontia and ectodermal dysplasias can be caused by reduced Wnt-β-catenin signaling due to pathogenic variants in WNT10A and LRP6.2, 25, 26, 27 In addition, pathogenic variants in EDA, a downstream target of Wnt/β-catenin signaling, cause X-linked ectodermal dysplasia28 and non-syndromic hypodontia.29 On the other hand, deletion of the WSC domain in KREMEN2 abolishes Dickkopf-1 binding and maintains Wnt signaling,11 which is consistent with the finding that protein-truncating variants in the negative Wnt regulator Axin-2 give rise to autosomal dominant hypodontia.30 It is possible that temporal and spatial variations in Dickkopf expression may result in KREMEN1 having roles as both an activator and an antagonist of Wnt signaling during tooth development.
Finally, differences in origin of isolated tooth agenesis versus syndromic oligodontia in families SO1 and SO2 are suggested by two observations. Isolated tooth agenesis does not segregate with either homozygosity or heterozygosity for KREMEN1 p.F209S. Furthermore, the teeth affected in family members with isolated tooth agenesis (upper lateral incisors and lower second premolars) are those generally encountered in sporadic hypodontia, which has multiple causes and is one of the most common human developmental anomalies.3, 31
In summary, our results strongly suggest that the genetics of KREMEN1 complement its cellular and physiological roles, just as do the genetics of WNT10A, LRP6, and EDA. The role of KREMEN1 in recessive ectodermal dysplasia expands the spectrum of the genetics of tooth agenesis.
References
Nieminen P : Genetic basis of tooth agenesis. J Exp Zool B Mol Dev Evol 2009; 312B: 320–342.
Massink MP, Créton MA, Spanevello F et al: Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am J Hum Genet 2015; 97: 621–626.
Arte S, Nieminen P, Apajalahti S, Haavikko K, Theslef I, Pirinen S : Characteristics of incisor-premolar hypodontia in families. J Dental Res 2001; 80: 1445–1450.
Visinoni AF, Lisboa-Costa T, Pagnan NA, Chautard-Freire-Maia EA : Ectodermal dysplasias: clinical and molecular review. Am J Med Genet Part A 2009; 149A: 1980–2002.
DeCoster PJ, Marks LA, Martens LC, Huysseune A : Dental agenesis: genetic and clinical perspectives. J Oral Pathol Med 2009; 38: 1–17.
Walsh T, Shahin H, Elkan-Miller T et al: Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of non-syndromic hearing loss DFNB82. Am J Hum Genet 2010; 87: 90–94.
Navon-Elkan P, Pierce SB, Segel R et al: Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014; 370: 921–931.
Pinheiro M, Freire-Maia N : Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet 1994; 53: 153–162.
Matalova E, Fleischmannova J, Sharpe PT, Tucker AS : Tooth agenesis: from molecular genetics to molecular dentistry. J Dental Res 2008; 87: 617–623.
Courtois G, Smahi A, Reichenbach J et al: A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest 2003; 112: 1108–1115.
Ponting CP, Hofmann K, Bork P : A latrophilin/CL-1-like GPS domain in polycystin-1. Curr Biol 1999; 9: R585–R588.
Nakamura T, Aoki S, Kitajima K, Takahashi T, Matsumoto K, Nakamura T : Molecular cloning and characterization of Kremen, a novel kringle-containing transmembrane protein. Biochim Biophys Acta 2001; 1518: 63–72.
Mao B, Wu W, Davidson G et al: Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signaling. Nature 2002; 417: 664–667.
Mao B, Wu W, Li Y et al: LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001; 411: 321–325.
Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C : Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998; 391: 357–362.
Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA : Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat Cell Biol 2001; 3: 683–686.
Mao B, Niehrs C : Kremen-2 modulates Dickkopf-2 activity during Wnt/LRP6 signaling. Gene 2003; 302: 179–183.
Hassler C, Cruciat CM, Huang YL, Kuriyama S, Mayor R, Niehrs C : Kremen is required for neural crest induction in Xenopus and promotes LRP6-mediated Wnt signaling. Development 2007; 134: 4255–4263.
Cselenyi CS, Lee E : context-dependent activation or inhibition of Wnt-beta-catenin signaling by Kremen. Science Signal 2008; 1: pe10.
Feng Q, Gao N : Keeping Wnt signalosome in check by vesicular traffic. J Cell Physiol 2015; 230: 1170–1180.
Andl T, Reddy ST, Gaddapara T, Millar SE : WNT signals are required for the initiation of hair follicle development. Dev Cell 2002; 2: 643–653.
Liu F, Chu EY, Watt B et al: Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol 2008; 313: 210–224.
Chen J, Lan Y, Baek JA, Gao Y, Jiang R : Wnt/beta-catenin signaling plays an essential role in activation of odontogenic mesenchyme during early tooth development. Dev Biol 2009; 334: 174–185.
Bohring A, Stamm T, Spaich C et al: WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 2009; 85: 97–105.
Adaimy L, Chouery E, Megarbane H et al: Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet 2007; 81: 821–828.
Cluzeau C, Hadj-Rabia S, Jambou M et al: Only four genes (EDA1, EDAR, EDARADD, and WNT10A account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mut 2011; 32: 70–72.
Kantaputra P, Sripathomsawat W : WNT10A and isolated hypodontia. Am J Med Genet Part A 2011; 155A: 1119–112.
Kere J, Srivastava AK, Montonen O et al: X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 1996; 13: 409–416.
Kurban M, Michailidis E, Wajid M, Shimomura Y, Christiano AM : A common founder mutation in the EDA-A1 gene in X-linked hypodontia. Dermatology 2010; 221: 243–247.
Lammi L, Arte S, Somer M et al: Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74: 1043–1050.
Vastardis H : The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop 2000; 117: 650–656.
Acknowledgements
We thank the families for their participation in this project and Ameer Hassouneh, DDS, for help in referral and dental examinations. This project was supported by the US Agency for International Development Program for Middle East Regional Cooperation award number TA-MOU10-M30-021 to Dr Kanaan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Issa, Y., Kamal, L., Rayyan, A. et al. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur J Hum Genet 24, 1430–1435 (2016). https://doi.org/10.1038/ejhg.2016.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.29
