
Abstract
Familial visceral myopathy (FVM) is a rare heritable and heterogeneous condition due to impaired smooth muscle function. We identified a family segregating 11 individuals with a spectrum of visceral symptoms involving the small intestine, colon, biliary tract, urinary tract and uterus. Whole-exome sequencing revealed a novel heterozygous tandem base substitution c.806_807delinsAA (p.(Gly269Glu)) in ACTG2, encoding smooth muscle actin γ-2, in affected family members. Variants in ACTG2 were recently identified in FVM with intestinal pseudo-obstruction as well as with the congenital megacystics-microcolon-intestinal hypoperistalsis syndrome. In our family, eight affected members presented with severe complications from the biliary and/or the urinary tracts in addition to gastrointestinal pseudo-obstructions. Furthermore, all affected mothers had a history of assisted deliveries owing to poor progress during labor and weak uterine contractions. The variable involvement of multiple smooth muscle-dependent organs in our family, including the biliary tract and the uterus, add to the phenotypic spectrum associated with ACTG2 missense variants.
Similar content being viewed by others

Introduction
Visceral myopathies (MIM: 155310 and 613834) comprise a group of heterogeneous genetic disorders owing to compromised function of the smooth muscle cells.1, 2, 3 The first genes associated with smooth muscle myopathy were MYH11 and ACTA2, both of which encode components of the contractile sarcomere, in families with thoracic aortic dissections (TAADs; MIM: 132900 and 611788).4, 5 Later, variants in ACTA2 were shown to be associated with multisystemic smooth muscle dysfunction syndrome (MIM: 613834) and a broad phenotypic spectrum.6, 7 A group among the visceral myopathies comprises the enteric visceral myopathies characterized predominantly by impaired gastrointestinal propulsion resulting in pain, distention, malabsorption and even death.8, 9 Reports on familial cases have indicated an autosomal dominant inheritance.10, 11 The clinical variability is extensive and a severe form is the megacystis-microcolon-intestinal hypoperistalsis (MMIH; MIM: 249210) syndrome with prenatal or neonatal onset of gastrointestinal obstruction and bladder enlargement.12, 13, 14, 15 In contrast, milder forms of enteric visceral myopathy may remain undiagnosed until adulthood.14, 16 Because of the variable symptomatology, this group of visceral myopathies constitutes a diagnostic challenge and some adult patients are referred to as chronic pseudo-obstruction, hollow visceral myopathy or pseudo-Hirschprung disease.2, 17
The findings on pathogenic MYH11 and ACTA2 variants in TAAD have raised the possibility that different components of the smooth muscle contractile unit and cytoskeleton may be responsible for other types of myopathies. Indeed, a recent study on a Finnish family segregating autosomal dominant familial visceral myopathy (FVM) with severe abdominal symptoms revealed segregation of a c.422C>A (p.(Arg148Ser)) variant in the ACTG2 gene.14 The gene encodes enteric smooth muscle actin γ-2 and the finding implied that the mechanism causing FVM is perturbed contractile function. This was confirmed by expression analysis of the c.422C>A variant showing reduced levels of the ACTG2 protein and a predicted poor actin polymerization. More recent studies identified several heterozygous missense variants in ACTG2 in MMIH patients that had occurred de novo or were inherited.15, 16 Notably, all disease-associated variants in ACTG2 identified to date predict missense substitutions and the absence of loss-of-function variants in affected individuals has led to the suggestion of a dominant-negative effect in FVM.16
We investigated a large Swedish family segregating FVM with symptoms from the intestines, the urinary tract and the biliary tract, as well as the uterus. Whole-exome sequencing (WES) revealed a novel heterozygous tandem base substitution (TBS) in ACTG2 in all affected family members and further analysis supported a dominant-negative mechanism of the mutated actin. The observations from our family add to the clinical spectrum associated with ACTG2 variants that may be considered in cases with variable symptoms from visceral organs.
Materials and methods
Patients
We investigated a Swedish family with 11 individuals affected by visceral symptoms consistent with autosomal dominant inheritance (Figure 1a). Detailed medical records were available from nine affected family members and seven of these were available for investigation and sampling. In two individuals (individuals (ind.) II:1 and ind. II:2), the diagnosis relied on anamnestic information from living relatives who reported on frequent hospitalizations, recurrent abdominal problems and death because of intestinal obstruction at age 18 and 38 years, respectively.

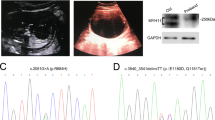
Pedigree of the Swedish family segregating FVM and a TBS in ACTG2. (a) The genotype at cDNA positions 806–807 of ACTG2 is shown below each symbol (GC, wild-type allele; AA, disease-associated allele). Affected individuals are shown with black filled symbols. Ind. I:2 with suspected disease is shown in gray. Asterisks denote subjects analyzed by WES. (b) Chromatogram of part of the ACTG2 transcript from fibroblasts in the affected ind. III:9 showing expression of the wild-type (GC) and the mutated transcripts (AA) at cDNA positions 806–807.
The clinical onset among affected members varied, and the first interactions with health care because of visceral symptoms were between one or a few months of age (ind. III:4, III:8, IV:2 and IV:4) until age 14–16 years (ind. II:4, II:7 and II:8; Table 1). Gastrointestinal symptoms were present and of variable degree in all 11 family members. Obstipation, distension, abdominal pain and diarrhea were the most common complaints. Seven affected members had undergone abdominal surgery, and malformations of the small intestine, cecum and colon (malrotation, widened jejunum and cecum, extended, thin or enlarged colon) were identified in six of these individuals. Abnormal intestinal peristalsis was confirmed in seven affected cases. Notably, the affected female (ind. III:2) had an overt clinical onset with cholelithiasis diagnosed at age 9 years. She was found to have enlarged bladder with residual urine and she suffered from recurrent upper urinary tract infections. Furthermore, she has one healthy child delivered by acute caesarian section because of poor progress in labor and impaired uterine contractions. Similarly, two additional affected females (ind. II:8 and III:9) who gave birth to altogether four children had a history of poor labor progress and assisted deliveries because of weak uterine contractions. One affected male (ind. III:4) had a clinical onset at 1 month of age because of urinary retention and enlarged bladder. As an adult, he was diagnosed with recurrent cholecystitis and impaired gastrointestinal peristalsis. Moreover, his father (ind. II:4) was diagnosed with biliary tract symptoms including cholecystitis and cholelithiasis in addition to gastrointestinal obstructions and enlarged bladder. Severe complications from the urinary tract were found in altogether seven affected family members and this consisted of enlarged bladder, residual urine and recurrent infections. No medical records or reliable clinical data were obtained for generation I, but information from ind. II:6 revealed that his mother (ind. I:2) had ‘recurrent stomach pain’, suggesting that she may have been affected. Informed written consent was obtained from all participating subjects or their legal guardians. This study was approved by the regional ethical committee of Uppsala, Sweden.
Genetic analysis
Whole-exome sequencing was conducted on DNA samples from four affected individuals (III:8, III:9, IV:2 and IV:4). The DNA was sheared using a Covaris instrument (Covaris Inc., Woburn, MA, USA). Fragment libraries were created from the sheared samples using AB Library Builder System and size selected on the BluePippin instrument (Sage Science, Beverly, MA, USA). Target enrichment was performed using the Agilent SureSelect Human All Exon 50 Mb Kit (Agilent, Santa Clara, CA, USA), according to the manufacturer’s protocols. Exome capture was conducted by hybridizing the DNA libraries with biotinylated RNA baits for 24 h followed by extraction using streptavidin-coated magnetic beads. Captured DNA was amplified by emulsion PCR using the Ion OneTouch 2 system and the Ion PI Template OT2 200 Kit (Life Technologies, Carlsbad, CA, USA) chemistry, followed by enrichment using Ion OneTouch ES. Samples were loaded on an Ion PI chip and sequenced on the Ion Proton System using Ion PI Sequencing 200 Kit (200 bp read length; Life Technologies). Alignment of reads to the human reference sequence (hg19 assembly) and variant detection was performed using plug-ins available in Torrent Suite software v.3.6 (Life Technologies). SNP and indel data was imported into a local installation of the CanvasDB system18 for storage and analysis of wholeexome variant data. Annotation information was obtained from ANNOVAR and dbSNP137. CanvasDB filtering analysis was performed to identify potentially damaging variants shared between the four patients while absent in any of the other ~900 exomes in our in-house database. Bidirectional Sanger sequencing (Applied Biosystems BigDye Terminator v.3.1 Cycle Sequencing Kit; Applied Biosystems, Waltham, MA, USA) on a 3730xl DNA Analyzer (Applied Biosystems) was used for segregation analysis of the identified variants as well as for the analysis of ACTG2 cDNA in affected family members and controls. Sequence analysis was performed using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA). Prediction of sequence variants on protein function was performed using PolyPhen-2 analysis19 and the NNSplice software (Berkley, CA, USA) from the Berkeley Drosophila Genome Project was used for analysis of possible changes of splice sites.20
ACTG2 transcript analysis
Total RNA was extracted from fibroblast cultures derived from the affected ind. III:8, III:9 and IV:2, as well as from three healthy unrelated individuals, using the PureLink RNA Mini Kit (Ambion by Life Technologies) according to the manufacturer’s instructions. cDNA synthesis was performed with the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). The ACTG2 primers were designed using Primer3Plus software.21 Analysis of a predicted cryptic splice site in exon 8 was performed using primers in exons 7 and 8. To test for possible exon skipping of exon 8, we size estimated and sequenced amplicons using primers in exons 7 and 9. The PCR products were size estimated on gels and sequenced. RT-qPCR was performed using FastStart Universal SYBR Green Master (ROX) (Roche, Basel, Switzerland) and normalized to β-actin. Primer sequences are available upon request. RT-qPCR experiments were carried out on StepOnePlus Real-Time PCR System (Applied Biosystems) using StepOne software v.2.2.2 (Life Technologies). Samples were run in triplicates and statistical analysis was performed using Student’s two-tailed t-test assuming equal variance.
Immunohistochemistry
Tissue samples for immunohistochemistry were obtained from distal ileum, colon, gall bladder, bile duct, ureter and uterus of anonymous healthy subjects, as well as from distal ileum from one affected family member (ind. IV:2).
All samples were fixed in 4% buffered formaldehyde, paraffin-embedded, sectioned (3 μm) and stained with hematoxylin and eosin (H&E) according to standard procedures. The tissue sections were deparaffinized in 100% xylene and rehydrated in decreasing concentrations of ethanol (100–80%) according to standard procedures. Sections were then heated in Tris-EDTA buffer (pH 9.0) (Dako, Glostrup, Denmark) for antigen retrieval and washed in distilled water. Staining was performed with primary rabbit anti-ACTG2 antibody (ab52219, 1:2000; Abcam, Cambridge, UK) for 30 min at room temperature in Tris-EDTA buffer (pH 9.0). Staining was visualized by the use of Dako REAL Envision Peroxidase/DAB detection system (Dako) and hematoxylin counterstaining. The sections were investigated in an Olympus BX51 microscope (Olympus, Hamburg, Germany) and photographs were taken using an Olympus SC30 camera and the Olympus software program cell Sense Entry 1.8 (Olympus, Tokyo, Japan).
3D modeling of the ACTG2
To understand the possible effect of the TBS on ACTG2, we compared the wild-type (wt) and mutated forms of ACTG2 in a 3D model. The model was based on the chick ACTG2 crystal structure with a protein sequence identical to that of human ACTG2. The Swiss model22 was used to create 14-mer models of the two protein variants and the CCP4mg software23 was used for 3D visualization of residue 269 in ACTG2, as well as its relative position to adjacent amino acids.
Results
The family history, medical records and clinical investigations revealed symptoms consistent with autosomal dominant FVM. Four affected family members were selected for WES and quality check of the aligned data set showed that 97% of the targeted exons were covered with at least 1 × on average, whereas 90% were covered by >20 ×. We then searched for shared variants in the four affected individuals and we identified seven heterozygous nonsynonymous nucleotide substitutions not present in dbSNP137. Subsequent segregation analysis using Sanger sequencing excluded six of the shared variants. However, all seven affected and sampled individuals were heterozygous for a TBS, c.806_807delinsAA, in ACTG2 (NM_001615.3; ClinVar database rs587777870 at NCBI; http://www.ncbi.nlm.nih.gov/clinvar/), whereas the three asymptomatic family members ‘at risk’ were non-carriers (Figure 1a). The TBSs are rare genomic variants and their involvement in human inherited disease was recently reviewed.24 The TBS in our family predicts a p.(Gly269Glu) substitution and the glycine residue at position 269 is highly conserved along the phylogenic scale (GERP score 4.05). Furthermore, the amino-acid substitution is predicted to affect protein function by PolyPhen-2 analysis (HumVar score 0.998, probably damaging).19 The variant was excluded in 1800 control chromosomes and it is not present in the EVS data release (ESP6500SI-V2) on the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) (Seattle, WA, USA; URL: http://evs.gs.washington.edu/EVS/) or the 63 352 unrelated exome-sequenced individuals available from the Exome Aggregation Consortium (ExAC) (Cambridge, MA, USA; URL: http://exac.broadinstitute.org, when accessed on October 2014). In combination with the previous reports on ACTG2 variants in FVM and MMIH, we thus considered the c.806_807delinsAA substitution a plausible cause of disease.
The two nucleotides c.806_807 in ACTG2 are located at the first two positions of exon 8 (exons numbered according to Ensembl transcript ENST00000345517; URL: http://www.ensembl.org) and the TBS predicts a loss of the acceptor splice site motif (exon|intron border wt: ctag|GCAT; mut: ctag.AAAT; NNSplice score: wt 0.37).
We then analyzed ACTG2 mRNA from fibroblast cells in three affected family members (III:8, III:9 and IV:2) and three healthy control individuals by RT-qPCR. Expression levels of ACTG2 in fibroblasts were low, yet detectable in all samples. The affected subjects showed a threefold reduction of ACTG2 expression when compared with controls (P<0.05, two-tailed t-test; Supplementary Figure 1a). To detect a possible skipping of exon 8 as well as activation of a predicted cryptic splice site in exon 8, we generated cDNA amplicons using primers in ACTG2 exons 7–9. The resulting RT-PCR products from patients and controls were of expected sizes without indications of shorter or truncated amplicons. Sanger sequencing of the amplicons confirmed that the c.806_807delinsAA substitution is present in a correctly spliced exon 8 (Figure 1b).
Immunohistochemical analysis of ACTG2 showed strong signals in smooth muscle cells of the small intestine, colon, bile duct, bladder, urethra and uterus from control individuals. A similar strong staining was observed in all muscle layers of a full biopsy from distal ileum and proximal cecum in one affected family member (IV:2) without detectable reductions in intensity when compared with a control specimen (Supplementary Figure 1b). Furthermore, the analysis did not reveal any fibrosis or tissue abnormalities using x20 magnification.
We then used the X-ray crystal structure of the chicken ACTG2 (PDB ID: P63270) to further predict the pathophysiological significance of the variant at a protein level. Our 3D model confirmed that residue 269 is positioned in a protruding loop of subdomain 4 of actin. In the wt actin polymer, the residue 269 is located in close proximity (7.49 and 3.91 Å, respectively) to residues 64 and 204 of the adjacent actin. However, these distances are changed when the uncharged glycine, which lacks a side chain, is replaced by the larger and polar glutamic acid with a relatively bulky side chain (Figure 2).

Structural protein model of wt and mutated ACTG2. Selected part of the ACTG2 model highlighting residue 269 in the actin monomer (light gray) and its relative position to the adjacent actin monomer (deep gray) in the actin polymer. The left panel shows the wt residue Gly269 in close proximity to residues 204, 64 and 60 of the neighboring actin monomer (3.91 Å from Thr204, 7.49 Å from Gly64 and 9.47 Å from Gln60, respectively). The right panel shows the missense variant Glu269 and its side chain that extends further out from the loop. Glutamic acid at position 269 alters distances (and charge) with respect to the closest residues in the neighboring actin monomer (4.44 Å from Thr204, 9.31 Å from Gln64 and 11.05 Å from Gln60, respectively), suggesting an effect on actin monomer interactions.
Discussion
The two smooth muscle actins ACTA2 and ACTG2 are linked to various phenotypes. Pathogenic variants in ACTA2 are predominantly associated with vascular symptoms including familial thoracic aneurysms,5 but may in rare cases involve visceral organs including the urinary and gastrointestinal tracts.6, 7 On the other hand, ACTG2 was recently found mutated in a family with FVM and adult gastrointestinal problems,14 as well as in the MMIH syndrome with severe neonatal symptoms from the intestines and the urinary tract.15, 16 The family investigated in this study shows a phenotypic overlap with both FVM and the MMIH syndrome but our observations on the variable onset as well as the involvement of the bile tract and uterus add to previous reports. We initially considered genetic heterogeneity and performed WES on four affected family members. The analysis revealed a heterozygous and novel GC to AA tandem substitution (c.806_807delinsAA) in ACTG2 segregating the disease. The TBSs, frequently designated as ‘small indels’, are rare and account for around 0.4% of all sequence variants described in the Human Gene Mutation Database (HGMD; http://www.hgmd.org).24, 25 Among the different TBSs, a change of CG to AA is the most prevalent type.24 The variant observed in our family results in a glycine to glutamic acid substitution (p.Gly269Glu) predicted to be damaging. In combination with previous reports on ACTG2 variants in visceral myopathies, our findings suggested the TBS c.806_807delinsAA as a likely cause of the disease.
The extensive clinical variability associated with ACTG2 variants from previous reports as well as within our family is intriguing. The first interactions with health-care units because of visceral symptoms varied from 1 month of age to 16 years of age in our study and, based on anamnestic information, possibly later in two deceased cases. All 11 affected individuals had a history of variable symptoms from the small intestine and colon. Obstipation, diarrhea, pain and distention were the most common complaints. Malformations and anatomical anomalies consisting of enlarged bladder, intestinal malrotation, and widened, extended or misplaced intestines were identified in six affected individuals. Urinary tract symptoms were evident in eight affected family members. In one individual (III:4) the initial symptom was urine retention.
In addition, three affected family members suffered from complications in the bile tract and, notably, one had an onset at 9 years of age. Secretion of bile into the intestinal lumen is dependent on smooth muscle contractions in the gall bladder. Thus, it may be hypothesized that inefficient or abolished propulsion of bile due to impaired smooth muscle contractions is a triggering factor for cholecystitis and inflammatory processes of the bile tract in our family. Additionally, we noted that the three affected mothers had given birth to a total of five children after lengthy labors. All deliveries were preceded by a poor labor progress that required assistance and, in one case, an acute caesarian section. The intensity and frequency of uterine contractions were low according to medical records and anamnestic information. Likewise, this suggests that impaired smooth muscle function resulting in inefficient and/or weak contractions of the uterus is a plausible reason for the complications during labor and deliveries in mothers with FVM.
Histopathological analysis of all muscle layers in distal ileum after intestinal resection in one affected member (ind. IV:2) showed a normal ACTG2 staining without degeneration or fibrosis of the muscle layers. Abnormal intracellular inclusions and slightly reduced ACTG2 staining have been reported in intestinal muscle layers of some FVM patients,14 but this was not observed in our case from the analysis of one specimen. The normal histology in intestinal smooth muscle layers of our patient may be related to her relatively young age, the possibility of focal abnormalities as well as the limited resolution from light microscopy. Sequencing of the RT-PCR products indicated that the mutated transcript was correctly spliced. Unexpectedly, qRT-PCR analysis of ACTG2 mRNA in cultured fibroblasts revealed reduced levels of the transcript in patients when compared with controls but the significance of this finding remains unclear. On the other hand, an effect of the TBS on actin structure was somewhat supported from our 3D modeling. The c.806_807delinsAA variant results in the replacement of the small and nonpolar glycine for the larger and polar glutamic acid (p.Gly269Glu). The residue 269 is located in close proximity to the neighboring actin monomer in the polymer structure. When replacing glycine for glutamic acid, the 3D model predicts an altered physical distance to the closest residues in the adjacent monomer. In combination with the different charges of the two amino acids, this suggests a possible effect on the actin polymer. Thus, it can be hypothesized that the substitution at position 269 perturbs polymerization and/or stability of the actin polymer, but this remains to be confirmed. In line with this, all previously reported ACTG2 variants associated with FVM and MMIH are missenses, suggesting a dominant-negative effect behind the disorders. Consequently, haploinsufficiency for ACTG2 may result in a clinically different or a silent phenotype. This is supported by the accidental finding of a nonsense variant c.187C>T (p.Arg63*) in ACTG2 in a single individual out of 1900 control subjects.16 The carrier of the truncating variant had mild constipation that did not require any medical treatment.
In summary, the novel ACTG2 TBS reported herein is associated with a broad spectrum of visceral symptoms involving the small intestine, colon, urinary tract, bile tract and uterus in 11 affected family members. The clinical expression showed a considerable variability, although gastrointestinal pseudo-obstruction was the most prevalent complication. Notably, symptoms from the bile tract and the uterus have not previously been associated with ACTG2 variants. Thus, our findings broaden the spectrum of clinical features in FVM of importance in the investigations and counseling of similar cases.
References
Lewis RA, Merin LM : Iris flocculi and familial aortic dissection. Arch Ophthalmol 1995; 113: 1330–1331.
Krishnamurthy S, Schuffler MD : Pathology of neuromuscular disorders of the small intestine and colon. Gastroenterology 1987; 93: 610–639.
Higman D, Peters P, Stewart M : Familial hollow visceral myopathy with varying urological manifestations. Br J Urol 1992; 70: 435–438.
Zhu L, Vranckx R, Khau Van Kien P et al: Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 2006; 38: 343–349.
Guo DC, Pannu H, Tran-Fadulu V et al: Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39: 1488–1493.
Richer J, Milewicz DM, Gow R et al: R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet Part A 2012; 158A: 664–668.
Milewicz DM, Ostergaard JR, Ala-Kokko LM et al: De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet Part A 2010; 152A: 2437–2443.
Moore SW, Schneider JW, Kaschula RO : Unusual variations of gastrointestinal smooth muscle abnormalities associated with chronic intestinal pseudo-obstruction. Pediatr Surg Int 2002; 18: 13–20.
Antonucci A, Fronzoni L, Cogliandro L et al: Chronic intestinal pseudo-obstruction. World J Gastroenterol 2008; 14: 2953–2961.
Jacobs E, Ardichvili D, Perissino A, Gottignies P, Hanssens JF : A case of familial visceral myopathy with atrophy and fibrosis of the longitudinal muscle layer of the entire small bowel. Gastroenterology 1979; 77: 745–750.
Rodrigues CA, Shepherd NA, Lennard-Jones JE, Hawley PR, Thompson HH : Familial visceral myopathy: a family with at least six involved members. Gut 1989; 30: 1285–1292.
Berdon WE, Baker DH, Blanc WA, Gay B, Santulli TV, Donovan C : Megacystis-microcolon-intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. Am J Roentgenol 1976; 126: 957–964.
Gosemann JH, Puri P : Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int 2011; 27: 1041–1046.
Lehtonen HJ, Sipponen T, Tojkander S et al: Segregation of a missense variant in enteric smooth muscle actin gamma-2 with autosomal dominant familial visceral myopathy. Gastroenterology 2012; 143: e1483.
Thorson W, Diaz-Horta O, Foster J II et al: De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet 2014; 133: 737–742.
Wangler MF, Gonzaga-Jauregui C, Gambin T et al: Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet 2014; 10: e1004258.
Di Lorenzo C, Youssef NN : Diagnosis and management of intestinal motility disorders. Semin Pediatr Surg 2010; 19: 50–58.
Ameur A, Bunikis I, Enroth S, Gyllensten U : CanvasDB: a local database infrastructure for analysis of targeted- and whole genome re-sequencing projects. Database (Oxford) 2014; 2014: 1–10.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Meth 2010; 7: 248–249.
Reese MG, Eeckman FH, Kulp D, Haussler D : Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Arnold K, Bordoli L, Kopp J, Schwede T : The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics (Oxford, England) 2006; 22: 195–201.
McNicholas S, Potterton E, Wilson KS, Noble ME : Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D 2011; 67: 386–394.
Chen JM, Ferec C, Cooper DN : Patterns and mutational signatures of tandem base substitutions causing human inherited disease. Hum Mutat 2013; 34: 1119–1130.
Chen JM, Cooper DN, Ferec C : A new and more accurate estimate of the rate of concurrent tandem-base substitution mutations in the human germline: approximately 0.4% of the single-nucleotide substitution mutation rate. Hum Mutat 2014; 35: 392–394.
Acknowledgements
We thank all family members for their cooperation and participation in the study as well as Dr Konstantin Tomanov, Max F Perutz Laboratories, Vienna, Austria, for valuable advice on 3D protein modeling. This work was supported by grants from the Swedish Research Council (K2013-66X-10829-20-3), Uppsala University Hospital, Uppsala University and the Science for Life Laboratory. JK was supported by the Swedish Society for Medical Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Klar, J., Raykova, D., Gustafson, E. et al. Phenotypic expansion of visceral myopathy associated with ACTG2 tandem base substitution. Eur J Hum Genet 23, 1679–1683 (2015). https://doi.org/10.1038/ejhg.2015.49
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.49
This article is cited by
-
Use of whole genome sequencing to determine the genetic basis of visceral myopathies including Prune Belly syndrome
Journal of Rare Diseases (2023)
-
Multi-disciplinary Insights from the First European Forum on Visceral Myopathy 2022 Meeting
Digestive Diseases and Sciences (2023)
-
Variants of the ACTG2 gene correlate with degree of severity and presence of megacystis in chronic intestinal pseudo-obstruction
European Journal of Human Genetics (2016)

{kind=link}