
Abstract
Brachydactyly type A1 is an autosomal dominant disorder primarily characterized by hypoplasia/aplasia of the middle phalanges of digits 2–5. Human and mouse genetic perturbations in the BMP-SMAD signaling pathway have been associated with many brachymesophalangies, including BDA1, as causative mutations in IHH and GDF5 have been previously identified. GDF5 interacts directly as the preferred ligand for the BMP type-1 receptor BMPR1B and is important for both chondrogenesis and digit formation. We report pathogenic variants in BMPR1B that are associated with complex BDA1. A c.975A>C (p.(Lys325Asn)) was identified in the first patient displaying absent middle phalanges and shortened distal phalanges of the toes in addition to the significant shortening of middle phalanges in digits 2, 3 and 5 of the hands. The second patient displayed a combination of brachydactyly and arachnodactyly. The sequencing of BMPR1B in this individual revealed a novel c.447-1G>A at a canonical acceptor splice site of exon 8, which is predicted to create a novel acceptor site, thus leading to a translational reading frameshift. Both mutations are most likely to act in a dominant-negative manner, similar to the effects observed in BMPR1B mutations that cause BDA2. These findings demonstrate that BMPR1B is another gene involved with the pathogenesis of BDA1 and illustrates the continuum of phenotypes between BDA1 and BDA2.
Similar content being viewed by others

Introduction
The brachydactylies constitute a collection of human digit phenotypes characterized by varying patterns of bone hypoplasia and malformed interphalangeal joints, which leads to shortened or absent tubular bones in the hands and/or feet. Genetic studies in humans and mice have revealed that most brachydactylies are attributed to perturbations in the bone morphogenetic protein (BMP) signaling pathway, specifically targeting the ligands, their antagonists and cognate receptors at the cell surface.1
Brachydactyly type A1 (BDA1, MIM #112500) is inherited as an autosomal dominant disorder and is primarily characterized by hypoplasia/aplasia of the middle phalanges of digits 2–5. Indian hedgehog (IHH) was the first gene associated with BDA12 and the mutations cluster in the central region of the N-terminal signaling fragment.3 Although the brachydactylies are isolated traits, there is considerable overlap in the pattern of the affected bones. For example, Ploger et al.4 described a BDA2 (MIM #112600) family where some of its affected members displayed shortened middle phalanges of digit 2, a classic BDA2 feature, along with various degrees of shortening of the middle phalanges of digits 3 and 4. Seemann et al.5 described another variation where in some patients the shortening of the middle phalange of digit 2, and, to a lesser extent digit 5, resembled Temtamy-type BDA4 (MIM %112800).6 The phenotypic overlap is suggestive of a common genetic pathway in phalangeal and metacarpal bone development. Work performed by Byrnes et al.7 and Degenkolbe et al.8 illustrated this point through the discovery of missense mutations in the growth differentiation factor 5 gene (GDF5) in patients diagnosed with BDA1. Dominant mutations in GDF5 were previously associated with four human conditions: that is, angel-shaped phalango-epihyseal dysplasia (ASPED; MIM 105835),9, 10 BDA2,4, 5, 11 BDC (MIM #113100),12, 13, 14, 15, 16 multiple synostosis (SYNS2; MIM #610017)8, 17 and symphalangism proximal 1B (SYM1B; MIM #615298),5, 18, 19 whereas severe chondrodysplasias of the Hunter–Thomson (MIM #201250),20 Grebe (MIM #200700),16, 21, 22 and Du Pan (MIM #228900)22 types were attributed to homozygous loss-of-function mutations.
The GDFs belong to the TGF-β superfamily of secretory signaling molecules that have diverse biological functions such as embryonic development and patterning, tissue homeostasis, immune response, reproduction and skeletal formation.23 GDF5 is a well-established osteo- and chondroinductive cytokine that preferentially binds with higher affinity to BMP receptor type-1B (BMPR1B) than to receptor type-1A (BMPR1A).24 These transmembrane serine-threonine kinase receptors belong to the TGF-β receptor superfamily. The mammalian BMP receptors are subclassified into 7 BMP type-1 receptors and 5 BMP type-2 receptors. BMPR1A and BMPR1B closely resemble the amino-acid composition of the activin receptor class 1 (ACVR1/ALK2), the gene responsible for fibrodysplasia ossificans progressive.25, 26 Phylogenetic analyses of the BMP type-1 receptor family suggest that both BMPR1B and BMPR1A co-evolved and are derived from the drosophila thickveins receptor (TKV),27 a receptor essential for visceral mesoderm patterning.28
Bmpr1a null mice are not viable, but mice carrying the inactivated Bmpr1a allele in chondrocytes exhibit hypoplasia of the long bones.29 In contrast, the skeletal defects of Bmpr1b null mice are restricted to the phalanges that display brachydactyly,29, 30, 31 similar to the GDF5 null mutant brachypodism mouse.32 Bmpr1b is the major transducer of BMP signals in early limb mesenchymal condensations.33 On ligand binding, heterotetrameric formation of BMP type-1 and BMP type-2 receptors occurs at the cell surface. This event triggers the intracellular transphosphorylation of the BMP type-1 receptor, which results in the phosphorylation of intracellular receptor-regulated SMADS causing it to translocate to the nucleus where it regulates transcriptional targets. Dominant mutations in the BMPR1B gene are associated with BDA2, BDC/SYM134, 35 and idiopathic pulmonary arterial hypertension (IPAH),36 whereas homozygous loss-of-function mutations cause acromesomelic chondrodysplasia.37, 38
We have a collection of BDA1 probands that do not carry mutations in either IHH or GDF5 and have been excluded for linkage to the BDA1 locus at chromosome 5p13.3 (BDA1B; MIM %607004). As GDF5 interacts directly with BMPR1B, we tested whether a subset of the probands in our cohort had mutations in the BMPR1B gene. We report the identification of two novel sequence variants in BMPR1B that are associated with BDA1.
Materials and methods
Ethics approval
This study was approved by the research ethics boards of the Ottawa Hospital and the Children's Hospital of Eastern Ontario. Genetic testing required voluntary informed consent by the patient or his/her legal guardians.
Clinical assessment
Individuals presenting with BDA1 features and who were previously found not to have BDA1-causing mutations in IHH and GDF5 were assessed for BDA1-causing mutations in the candidate gene BMPR1B. Metacarpophalangeal pattern profiles (MCPP) were performed on standardized hand radiographs by SN. The length of each hand bone, excluding the wrist bones, was measured using the software ANTRO39 and adjusted for age and sex as described previously.40 Tubular hand bones with a SD≤−2 were classified as being disproportionately short, whereas those with a SD≥+2 indicated disproportionately long bones.
Molecular analyses
Genomic DNA (gDNA) was extracted and column purified from whole blood using QIA Blood Mini Kit (Qiagen, Valencia, CA, USA) as per the manufacturer's protocol. Purified gDNA was amplified with primer pairs specific to each exon of BMPR1B. Each primer pair was designed by ExonPrimer to flank at least 50 bases from each intron–exon boundary as listed in Supplementary Table S1. PCR of each primer pair was performed in a 50-μl reaction as follows: 50 ng template, 10 μ M forward and reverse primer, 10 mM dNTPs, 2.0 mM MgCl2, 10 × PCR buffer and 1U rTaq (Sigma-Aldrich, St Louis, MO, USA). Cycling conditions were set at 95 °C 5 min initial denaturation, followed by 30 cycles of 95 °C 45 s denaturation, 55 °C 30 s annealing, 72 °C 30 s extension and a final extension at 72 °C for 10 min. PCR aliquots were analyzed on a 2% agarose gel and stained with 5 mg/ml EtBr before purification with AMPure XP (Agencourt, Beverly, MA, USA) on a Biomek FX automated workstation (Beckman Coulter, Brea, CA, USA). Samples were prepared for BigDyeV3.1 terminator sequencing on an AB 3730xl genetic analyzer (Life Technologies, Carlsbad, CA, USA) as previously described7 with the exception of the removal of unincorporated label by CleanSeq SPRI magnetic beads (Agencourt).
In-silico analyses
Each sequence was aligned to the human reference HG19 using BLAT to identify any sequence mismatches. Novel sequence variants were defined as having passed the filtering process through dbSNP138, Genome Variants, HGMD and EVS. Sequence variants meeting this criterion were evaluated for pathogenicity using PolyPhen-2 V2.2.2 and SIFT. Sequence variants that aligned within annotated splice sites were evaluated with online web services Human Splice Finder V2.4.1, SplicePort and Alternative Splice Site Prediction. RefSeq NM_001203.2 was used as input sequence for all of the prediction services. Sequence variants were deposited at http://databases.LOVD.nl/. Multiple sequence alignment of BMPR1B from human (AAH47773.1), mouse (AAH65106.1), cow (NP_001098798.1), sheep (NP_001009431.1), chicken (NP_990463.1), frog (NP_001072633.1), zebrafish (NP_571532.1) and drosophila TKV (AAA53243.1) was performed using ClustalOmega with default settings. Cn3D v4.3 was used to visualize the location of BMPR1B mutations on the crystal structure of BMPR1B (PDB ID: 3MDY_A). VAST was used to align structure neighbors (PDB IDs: 4C02_A and 1B6C_B).
Reporter constructs and cell culture
Assessment of cartilage-stimulating activity of wild-type and mutant BMPR1B constructs was assayed in limb bud-derived mesenchymal cultures as previously described.41 In brief, fore and hind limb buds were harvested from embryonic age 11.5 embryos, and the limb buds were dissociated and mesenchymal cells isolated. Transfection mixtures were prepared in FuGENE6 (Promega, Madison, WI, USA) with a 1:3 ratio of Sox responsive Col2-Luc reporter (+ phRL) to expression vector (control pKS, BMPR1BWT or BMPR1BK325N). One half-volume of transfection mixture was added to one volume of cells resuspended at a density of 2 × 107 cells/ml, and 10 microliters of this mixture was used to establish a micromass colony in the well of a 24-well tissue culture plate. One day post cultures, select wells were treated with BMP4 (R&D Systems, Minneapolis, MN, USA) to a final concentration of 20 ng/ml. Cell lysates were harvested 48 h post-transfection, and firefly and Renilla luciferase activity was measured using the Dual-Luciferase Reporter Assay kit (Promega). Renilla luciferase activity was used to normalize for transfection efficiency across samples.
Results
Family-1
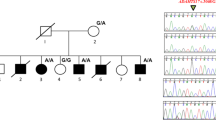
The proband was a well, developmentally normal 16-month-old Caucasian female, the first child born to a pair of healthy and unrelated parents (Figure 1a). No complications were noted during the pregnancy and she was born at term with a birth weight of 3410 g and length of 52 cm. Both parents appeared normal and did not exhibit any hand or foot anomalies. The proband’s stature was average (height 75th %ile, head circumference 50th%ile), but her index fingers were very short and the fifth fingers were slightly short with ulnar curvature (Supplementary Figure S1A). The palmar creases were normal but digital creases were reduced at the distal interphalangeal joints. Examination of the feet revealed shortened big toes. The second toes were very long with moderate syndactyly between toes 2 and 3. Hand radiographs showed a slightly reduced size of the terminal phalanx of the thumbs (Figure 1a). The index fingers lacked middle phalanges and the fifth digits had tiny middle phalanges. The MCPP revealed the presence of a short distal and proximal phalange of both thumbs, a shortening of the terminal and middle phalanges of digit 3, severe shortening of the middle phalanges of digit 2 and a slight shortening of the middle phalanges of digit 5 (Figure 1a). The bones that make up digit 4 appeared to be normal. Cytogenetic analysis revealed a normal karyotype.

Two novel BMPR1B mutations are associated with BDA1. (a, b) Assessment of Family-1. (a) A hand radiograph and MCPP profile of the proband (arrow) are illustrated to the right of the family trio. (b) Sequence trace reveals a heterozygous c.975A>C (p.(K325N)) in the proband. (c–f) Assessment of Family-2. (c) A hand radiograph and MCPP profile of the proband are displayed to the right of the family trio. (d) Sequence trace reveals a heterozygous c.447-1G>A in the proband. IUPAC code R denotes an A or G nucleotide base. (e, f) The sequence variant is predicted to splice into exon 8 at c.447, which would result in a shift in the translational reading frame.
Family-2
The proband who was of African descent, born to healthy and unrelated parents (Figure 1c) was initially seen at 11 months of age. The child sat, unsupported at 7 months, stood with supports at 11 months and walked at 14 months of age. Mild developmental delay was noted with speech and toileting delay, poor motor coordination and brachydactyly of the hands. All fingernails were present and there was no facial dysmorphology. A younger brother who had normal hands and feet also had speech delay. By age 9 years, the proband was doing well at school with extra support, had good hand function and was not dysmorphic. A hand radiograph taken from the proband displayed small cube-shaped middle phalanges of digit 2 and small trapezoid-shaped middle phalanges of digit 5 in both hands (Figure 1c). The middle phalange of digit 2 displayed a socket-like groove for the epiphysis of the proximal phalange. Clinodactyly in both the fifth digit and the right thumb was observed in the proband. There were no remarkable hand bone features in the parents (Supplementary Figure S1B). The MCPP from both hands revealed moderate shortening of the distal phalanges of digits 1 and 2, shortened proximal phalange of digit 1 and severe shortening of the middle phalanges of digits 2 and 5 (Figure 1c). The proband had a normal karyotype and was excluded for Fragile X.
Molecular analyses
The 16-month-old female patient in Family 1 did not carry a mutation in either IHH or GDF5. Sequencing of the BMPR1B gene revealed a novel heterozygous c.975A>C p.(Lys325Asn) in her (Figure 1b), but the variant was not found in the mother. The biological father was reported to have no hand or feet anomalies, but was unavailable to study. The sequence variant was not identified in any of the public variant databases including dbSNP138, Personal Genomes HGMD and Exome Variant Server. Moreover, we sequenced 100 controls and did not identify this variant in exon 10 of BMPR1B. The asparagine substitution at residue position 325 was predicted by both SIFT (score=0) and PolyPhen-2 (score=1) to be pathogenic. A ClustalOmega multiple alignment of the peptide sequence of BMPR1B from human, mouse, chicken and sheep positioned Lys325 within a highly conserved region of the cytoplasmic protein kinase domain (Figure 2a).

BMPR1B mutation spectrum. (a) The p.(K325N) substitution is located within a highly conserved region of the protein kinase domain of BMPR1B. (b) With the exception of two mutations that are associated with acromesomelic chondrodysplasia, mutations in BMPR1B are found in the intracellular signaling region. BDA1 (yellow), BDA2 and BDC/SYM1 (purple), acromesomelic chondrodysplasia (blue) and IPAH (orange). The BDA1 mutation lies within a VAST conserved β-strand. Transmembrane domain (TM). TGF-GS (Transforming growth factor β GS domain). (c) A Cn3D (v4.3) tube view of the intracellular region of BMPR1B (PDB ID: 3MDY_A). Secondary structures are colored as follows: alpha helices in green and beta strands in brown. Amino-acid substitutions are described next to each colored ball for a specific human condition; yellow represents BDA1 (p.(K325N)), purple is for BDA2 (p.I200P, p.R486Q and p.R486W) and BDC/SYM1 (p.R486Q), and orange is associated with IPAH (p.F392L).
We also report on an individual of African descent with a mix BDA1-arachnodactyly condition. All three family members did not have a mutation in either IHH or GDF5. The sequencing of BMPR1B revealed a heterozygous c.447-1G>A (HG19 NC000004.11:g.96046133G>A) in the proband (Figure 1d) and mother. The variant was not observed in 100 sequenced controls nor in any public variant databases. The nucleotide substitution occurred in a constitutive acceptor splice site for exon 8 of BMPR1B where the first two nucleotides AG are highly conserved in the consensus acceptor splice site sequence YYTT(Y)6NCAG│G.42 The G to A transition was predicted to abolish the 3′ splice site and to create a novel 3′ site just one nucleotide upstream at position c.447 (Figure 1e and Supplementary Table S2). This would result in a translational frameshift of one nucleotide that in turn produced a truncated peptide of 162 amino acids (Figure 1f).The two novel sequence variants in BMPR1B were deposited at the LOVD database with LOVD ID numbers 0000053136 (c.9775A>C) and 00029686 (c.447-1G>A)
Mutation distribution of BMPR1B
BMPR1B encodes for a 502 amino-acid protein with three main functional domains: namely, an extracellular ligand binding domain, a single transmembrane domain and an intracellular protein kinase domain. We found that the majority of mutations in BMPR1B resided within the protein kinase domain (Figure 2b). The superimposition of missense mutations onto the 3D model of BMPR1B suggests that mutations that are associated with BDA1 and BDA2 are found on the outer edges of the cytoplasmic region of BMPR1B, whereas the mutation that is associated with IPAH is buried within four alpha helices (Figure 2c). In addition, a VAST structure alignment of closely related cytoplasmic peptide chains revealed a conserved stretch of 23 residues (Figure 2b), which corresponded to an outward facing beta strand where the predicted p.(Lys325Asn) happens to be located (Figure 2c).
BMPR1BLys325Asn acts as a dominant-negative mutation
To assess the impact of c.975A>C p.(Lys325Asn) on BMPR1B activity, we measured the activity of this construct in comparison with wild-type BMPR1B in a BMP-responsive chondrogenic cell assay. Primary limb mesenchymal cells are highly responsive to BMP4 and other BMPs.43, 44 Under these conditions, addition of BMP2 or 4, or heterologous expression of constitutively activated BMPR1A or B receptors leads to pronounced induction of a chondrogenic-responsive reporter gene.44, 45, 46 In this assay, expression of BMPR1BLys325Asn led to an ~50% reduction in reporter gene activity when compared with wild-type BMPR1B (Figure 3). Moreover, we observed an ~60% reduction in reporter activity in response to BMP4, a BMPR1B ligand and an inducer of chondrogenesis.

BMPR1BK325N impacts receptor activity. Transfection of primary limb bud-derived mesenchymal cells was used to evaluate the impact of heterologous expression of either wildtype (BMPR1BWT), mutant (BMPR1BK325N), or control pKS on Sox-driven reporter activity (Col2-Luc). BMP4 (20 ng/ml) was added 24 h post transfection, and luciferase activity was assessed 48 h post transfection. P-values were calculated using one-way ANOVA with Tukey post-hoc test. Error bars represent 1 SD.
Discussion
Mutations in either IHH or GDF5 have been previously shown to be associated with BDA1.2, 3, 7 In this study we describe two novel sequence variants in the human BMPR1B gene that are associated with BDA1-like conditions in two unrelated individuals. In the first BDA1 case, a c.975A>C is predicted to cause a p.(Lys325Asn) substitution next to the serine-threonine kinase active site in the C-terminal domain of BMPR1B. While c.975A>C was predicted to be pathogenic, the expressed mutant demonstrated a significant reduction of reporter activity in the presence of BMP4. It is possible that the exchange of a positively charged amino acid like lysine for an uncharged asparagine residue could result in a loss of BMPR1B kinase activity, similar to the effect observed in the study describing the BDA2 mutation p.Ile200Lys.35 However, in the same study a recombinant chicken Bmpr1b construct containing the BDA2 mutation p.Arg486Trp was found to have normal kinase activity despite its strong inhibitory effect on chondrogenesis in micromass cultures. The serine-threonine domain of BMP type-1 receptors serves two purposes in establishing the proper intracellular response on ligand binding: (1) phosphorylation of the R-SMADs, and (2) proper complex formation between type-1 and type-2 receptors.47, 48 Thus, an alternative pathogenic mechanism to consider is the disruption of the heterotetrameric complex. The BDA1-associated variant c.975A>C (p.(Lys325Asn)) was located in a highly conserved structural motif (Figure 2b), which could be involved with receptor complex formation. The proper formation of the complex has been shown to affect the avidity for ligands,49 and it is possible that p.(Lys325Asn) could disrupt the strength of ligand binding to BMPR1B and BMPR1A. Recently, it has been shown that in vitro SMAD reporter assays of BDA1 mutations in GDF5, p.Arg399Cys and p.Trp414Arg, displayed a more severe reduction in BMPR1A signal as compared with BMPR1B.8 The authors suggested that a disruptive BMPR1A signaling could be the key pathogenic feature involved with causing BDA1. An evaluation of the effects of p.(Lys325Asn) in SMAD signaling could further support their model.
The second BDA1 case carried a novel heterozygous c.447-1G>A within a canonical acceptor splice site of exon 8 of BMPR1B. Mutations within acceptor splice sites have been previously shown to cause exon skipping and intron retention, which could translate into a truncated peptide.50 The predicted consequence of c.447-1G>A on the splicing of BMPR1B pre-mRNA would be the creation of an adjacent acceptor splice site at c.447. As the resulting transcript contains 13 additional codons before premature termination (Figure 1f), it could be subjected to nonsense mediated decay.51 If this is the case, then the BDA1 condition of the proband would be a result of haploinsufficiency of BMPR1B. Alternatively c.447-1G>A could act as a hypomorphic allele similar to what was described in a mouse mutant Ali30 carrying a T>G transversion in the splice donor site of exon 10 of Bmpr1b.52 These mutant mice exhibited brachydactyly in addition to retinopathy. Although the mother carried the c.447-1G>A variant, she did not exhibit the brachydactyly features observed in her son, indicative of reduced penetrance. It is possible that the environment and/or genes other than IHH and GDF5 could have modified the expression. Interestingly, the proband of family 2 exhibited restricted arachnodactyly features. Contractual arachnodactyly combined with brachydactyly and sensoneural hearing loss with no known mutation has been reported in a case study.53 Our patient’s hearing was unremarkable, and there were no indications of visual impairment as seen in the mouse mutant Ali30. It is unclear if the splice sequence variant in this family is contributing to the disproportionate lengthening of digits 3 and 4. Further functional characterization is needed to address this concern.
We did note the remarkable similarity in MCPP of both probands. There are also phenotypic similarities between our patient and the ones described with BDA2-causing mutations in BMPR1B. Our patient with the c.975A>C has a similar missing middle phalange of digit 2 as does the BDA2 patient with the p.Ile200Lys mutation. Unlike the reported BDA2 cases, both of our patients displayed the shortening of the proximal and distal phalange of the thumb, which is also distinct from BDC. These differences could be attributed to the position of the mutations within BMPR1B, similar to what has been observed in GDF5 for BDA1, BDA1/SYNS2 and BDA2.8
In summary, we identified two novel sequence variants in BMPR1B, which are associated with BDA1-like phenotypes. Their effects are predicted to disrupt SMAD signaling either in a dominant-negative manner (c.975A>C) or by haploinsufficiency (c.447-1G>A). Hence, our study further supports the role of the BMP-SMAD pathway in brachymesophalangies and partially reveals the nature of structurally conserved domains of BMPR1B.
References
Stricker S, Mundlos S : Mechanisms of digit formation: human malformation syndromes tell the story. Dev Dynamics 2011; 240: 990–1004.
Gao B, Hu J, Stricker S et al: A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range. Nature 2009; 458: 1196–1200.
Byrnes AM, Racacho L, Grimsey A et al: Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur J Hum Gen 2009; 17: 1112–1120.
Ploger F, Seemann P, Schmidt-von Kegler M et al: Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum Mol Genet 2008; 17: 1222–1233.
Seemann P, Schwappacher R, Kjaer KW et al: Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest 2005; 115: 2373–2381.
Temtamy SA, McKusick VA : The genetics of hand malformations. Birth Defect Orig Artic Ser 1978; 14: i–xviii, 1-619.
Byrnes AM, Racacho L, Nikkel SM et al: Mutations in GDF5 presenting as semidominant brachydactyly A1. Hum Mut 2010; 31: 1155–1162.
Degenkolbe E, Konig J, Zimmer J et al: A GDF5 point mutation strikes twice—causing BDA1 and SYNS2. PLoS Genet 2013; 9: e1003846.
Gutierrez-Amavizca BE, Brambila-Tapia AJ, Juarez-Vazquez CI et al: A novel mutation in CDMP1 causes brachydactyly type C with ‘angel-shaped phalanx’. A genotype-phenotype correlation in the mutational spectrum. Eur J Med Genet 2012; 55: 611–614.
Holder-Espinasse M, Escande F, Mayrargue E et al: Angel shaped phalangeal dysplasia, hip dysplasia, and positional teeth abnormalities are part of the brachydactyly C spectrum associated with CDMP-1 mutations. J Med Genet 2004; 41: e78.
Kjaer KW, Eiberg H, Hansen L et al: A mutation in the receptor binding site of GDF5 causes Mohr-Wriedt brachydactyly type A2. J Med Genet 2006; 43: 225–231.
Everman DB, Bartels CF, Yang Y et al: The mutational spectrum of brachydactyly type C. Am J Med Genet 2002; 112: 291–296.
Polinkovsky A, Robin NH, Thomas JT et al: Mutations in CDMP1 cause autosomal dominant brachydactyly type C. Nat Genet 1997; 17: 18–19.
Savarirayan R, White SM, Goodman FR et al: Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am J Med Genet Part A 2003; 117A: 136–142.
Schwabe GC, Turkmen S, Leschik G et al: Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. Am J Med Genet Part A 2004; 124A: 356–363.
Thomas JT, Kilpatrick MW, Lin K et al: Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 1997; 17: 58–64.
Dawson K, Seeman P, Sebald E et al: GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet 2006; 78: 708–712.
Wang X, Xiao F, Yang Q et al: A novel mutation in GDF5 causes autosomal dominant symphalangism in two Chinese families. Am J Med Genet Part A 2006; 140A: 1846–1853.
Yang W, Cao L, Liu W et al: Novel point mutations in GDF5 associated with two distinct limb malformations in Chinese: brachydactyly type C and proximal symphalangism. J Hum Genet 2008; 53: 368–374.
Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP : A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet 1996; 12: 315–317.
Al-Yahyaee SA, Al-Kindi MN, Habbal O, Kumar DS : Clinical and molecular analysis of Grebe acromesomelic dysplasia in an Omani family. Am J Med Genet Part A 2003; 121A: 9–14.
Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH et al: Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1 gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome). Clin Genet 2002; 61: 454–458.
Massague J : TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012; 13: 616–630.
Nickel J, Kotzsch A, Sebald W, Mueller TD : A single residue of GDF-5 defines binding specificity to BMP receptor IB. J Mol Biol 2005; 349: 933–947.
Shore EM, Xu M, Feldman GJ et al: A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38: 525–527.
Kaplan FS, Xu M, Seemann P et al: Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mut 2009; 30: 379–390.
Schmierer B, Hill CS : TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 2007; 8: 970–982.
Affolter M, Nellen D, Nussbaumer U, Basler K : Multiple requirements for the receptor serine/threonine kinase thick veins reveal novel functions of TGF beta homologs during Drosophila embryogenesis. Development 1994; 120: 3105–3117.
Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM : Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA 2005; 102: 5062–5067.
Baur ST, Mai JJ, Dymecki SM : Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. Development 2000; 127: 605–619.
Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM : The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 2000; 127: 621–630.
Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ : Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 1994; 368: 639–643.
Zou H, Wieser R, Massague J, Niswander L : Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Gene Dev 1997; 11: 2191–2203.
Lehmann K, Seemann P, Boergermann J et al: A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type C/symphalangism-like phenotype or brachydactyly type A2. Eur J Hum Genet 2006; 14: 1248–1254.
Lehmann K, Seemann P, Stricker S et al: Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci USA 2003; 100: 12277–12282.
Chida A, Shintani M, Nakayama T et al: Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J 2012; 76: 1501–1508.
Demirhan O, Turkmen S, Schwabe GC et al: A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J Med Genet 2005; 42: 314–317.
Graul-Neumann LM, Deichsel A, Wille U et al: Homozygous missense and nonsense mutations in BMPR1B cause acromesomelic chondrodysplasia-type Grebe. Eur J Hum Genet 2014; 22: 726–733.
Hosenfeld D, Hosenfeld F, Schaefer E, Grote W : IBM-PC compatible software for establishing metacarpophalangeal pattern profiles. Clin Genet 1991; 39: 396–400.
Armour CM, Bulman DE, Hunter AG : Clinical and radiological assessment of a family with mild brachydactyly type A1: the usefulness of metacarpophalangeal profiles. J Med Genet 2000; 37: 292–296.
Underhill TM, Dranse HJ, Hoffman LM : Analysis of chondrogenesis using micromass cultures of limb mesenchyme. Method Mol Biol 2014; 1130: 251–265.
Senapathy P, Shapiro MB, Harris NL : Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Method Enzymol 1990; 183: 252–278.
Barna M, Niswander L : Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev Cell 2007; 12: 931–941.
Hoffman LM, Garcha K, Karamboulas K et al: BMP action in skeletogenesis involves attenuation of retinoid signaling. J Cell Biol 2006; 174: 101–113.
Karamboulas K, Dranse HJ, Underhill TM : Regulation of BMP-dependent chondrogenesis in early limb mesenchyme by TGFbeta signals. J Cell Sci 2010; 123: 2068–2076.
Weston AD, Rosen V, Chandraratna RA, Underhill TM : Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J Cell Biol 2000; 148: 679–690.
Ehrlich M, Gutman O, Knaus P, Henis YI : Oligomeric interactions of TGF-beta and BMP receptors. FEBS Lett 2012; 586: 1885–1896.
Nohe A, Hassel S, Ehrlich M et al: The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 2002; 277: 5330–5338.
Sebald W, Nickel J, Zhang JL, Mueller TD : Molecular recognition in bone morphogenetic protein (BMP)/receptor interaction. Biol Chem 2004; 385: 697–710.
Licatalosi DD, Darnell RB : RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 2010; 11: 75–87.
Maquat LE : Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 2004; 5: 89–99.
Graw J, Puk O, Weber S et al. Bmpr1b splice site mutation leads to optic nerve head degeneration in the mouse. Invest Opthalmol Vis Sci 2012; Vol 53.
Alptekin N, Ozturk EA, Gok H, Yalcin P, Tekin M : Congenital contractural arachnodactyly, brachydactyly and sensorineural hearing loss: an unusual association. J Genet Couns 2005; 16: 421–423.
Acknowledgements
We thank the families for their participation. Funding was provided by an operating grant (DEB) from the Canadian Institute of Health Research. Online resources: ASSP: wangcomputing.com/assp/. ClustalOmega: www.ebi.ac.uk/Tools/msa/clustalo/. ExomeVariant Server: evs.gs.washington.edu/. HGMD: www.hgmd.org/. HumanSpliceFinder: www.umd.be/HSF/. PolyPhen-2: genetics.bwh.harvard.edu/pph2/. SIFT: sift.jcvi.org/. SplicePort: spliceport.cbcb.umd.edu/. VAST: http://structure.ncbi.nlm.nih.gov/Structure/VAST/.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Racacho, L., Byrnes, A., MacDonald, H. et al. Two novel disease-causing variants in BMPR1B are associated with brachydactyly type A1. Eur J Hum Genet 23, 1640–1645 (2015). https://doi.org/10.1038/ejhg.2015.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.38
This article is cited by
-
A novel variant in the ROR2 gene underlying brachydactyly type B: a case report
BMC Pediatrics (2022)
-
Complex Phenotypes: Mechanisms Underlying Variation in Human Stature
Current Osteoporosis Reports (2019)
-
Linked homozygous BMPR1B and PDHA2 variants in a consanguineous family with complex digit malformation and male infertility
European Journal of Human Genetics (2018)
-
Genome-wide association analyses identify 39 new susceptibility loci for diverticular disease
Nature Genetics (2018)
-
Mutant GDF5 enhances ameloblast differentiation via accelerated BMP2-induced Smad1/5/8 phosphorylation
Scientific Reports (2016)
