
Abstract
Mutations of FLNA, an X-linked gene that encodes the cytoskeletal protein filamin A, cause diverse and distinct phenotypes including periventricular nodular heterotopia and otopalatodigital spectrum disorders (OPDS). Craniofacial abnormalities associated with OPDS include supraorbital hyperostosis, down-slanting palpebral fissures and micrognathia; craniosynostosis was previously described in association with FLNA mutations in two individual case reports. Here we present four further OPDS subjects who have pathological FLNA variants and craniosynostosis, supporting a causal link. Together with the previously reported patients, frontometaphyseal dysplasia was the most common clinical diagnosis (four of six cases overall); five patients had multiple suture synostosis with the sagittal suture being the most frequently involved (also five patients). No genotype–phenotype correlation was evident in the distribution of FLNA mutations. This report highlights the need to consider a filaminopathy in the differential diagnosis of craniosynostosis, especially in the presence of atypical cranial or skeletal features.
Similar content being viewed by others


Introduction
The phenotypic spectrum associated with mutations in FLNA is unusually diverse and correlates with the functional consequence for the filamin A protein. Loss-of-function mutations were originally described in periventricular nodular heterotopia,1 a central nervous system migration defect; an extended phenotype is increasingly recognised in association with these mutations, which may include intestinal pseudo-obstruction/congenital short bowel syndrome,2 cardiac valvulopathies,3 vascular dilatation, joint hypermobility, and various skin manifestations.4
By contrast, gain-of-function FLNA mutations are characteristic of the skeletal and connective tissue disorders collectively termed otopalatodigital spectrum disorders (OPDS), which manifest a broad range of clinical severity.5 The mildest phenotype is otopalatodigital syndrome type 1 (OPD1), which presents in affected males with cleft palate, hearing loss (which can be conductive, sensorineural, or mixed), and a characteristic craniofacial phenotype of downslanting palpebral fissures and micrognathia; skeletally there are overlapping fingers and bowed long bones.6 These characteristics are also present in OPD type 2 (OPD2), a more severe phenotype associated with perinatal male lethality and additional midline defects such as hypospadias and omphalocele.5, 7, 8 Females who are obligate carriers of mutations causing both OPD1 and OPD2 have a variable, milder expression of similar characteristics.7, 8 Frontometaphyseal dysplasia (FMD) is described in both male and female subjects and presents with the cardinal features of prominent supraorbital ridges, hypertelorism, down-slanting palpebral fissures, and a generalised skeletal dysplasia that manifests with thickening of the calvarium, agenesis of the frontal, ethmoidal and sphenoidal sinuses, and bowing and undermodelling of the diaphyses and metaphyses of tubular bones. Other extra-skeletal manifestations include cardiac defects, laryngeal stenosis, and ureteric and urethral stenosis.9 Melnick–Needles syndrome (MNS) is usually lethal in males; facial features in affected females include prominent supraorbital ridge, exorbitism, oligohypodontia, and micrognathia, which are associated with multiple skeletal abnormalities that include late-closing fontanelles, skull base sclerosis, sinuous clavicles, irregular rib contour, bowed long bones, pelvic hypoplasia and elongated metacarpals.10 Terminal osseous dysplasia is lethal in males and manifests in female infancy with skeletal dysplasia particularly affecting the extremities, digital fibromata, abnormal facial and scalp skin pigmentation, and facial dysmorphism including hypertelorism, upslanting palpebral fissures, and epicanthic folds.11
Although craniofacial abnormalities across the OPDS are often a defining feature in clinical diagnosis, overt craniosynostosis (premature fusion of the cranial sutures) has only been reported in two separate mutation-proven cases.9, 10 Here we present four further subjects with craniosynostosis (two male and two female), who have documented FLNA variants. This novel association is important to recognise in the assessment of a patient with craniosynostosis.
Methods
DNA was extracted from peripheral blood of each patient and referred for diagnostic sequencing of the FLNA gene. Details of the primers used can be obtained from either of the two laboratories that undertook the testing (Genetics Laboratories, Oxford, UK or Clinical Genetics Group, University of Otago, New Zealand).
Results
Case reports
Patient 1
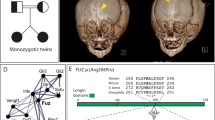
This female subject is the only child of unrelated parents; she was born at term via normal vaginal delivery after an uneventful pregnancy. She was referred for clinical genetic assessment at an age of 11 years because of dysmorphic features, difficulty with concentration and bilateral moderate–severe mixed hearing loss. On examination she had an irregular retroclined forehead and a prominent supraorbital ridge; there was mild hypertelorism and micrognathia but no substantial midface hypoplasia (Figure 1a and b). She had long slender fingers and toes, absent flexion of the interphalangeal joint of the thumbs, flexion deformity at the elbow, genu valgum, and bilateral hallux valgum. Radiographs showed undermodelling of the short tubular bones of the hand, bowing of the proximal radii, valgus bowing of the tibia, and wavy clavicles. Three-dimensional computed tomography (3D CT) scanning showed pansynostosis, a copper-beaten appearance, significant frontal bone thickening, and a strikingly prominent supraorbital rim (Figure 1c); intracranial pressure (ICP) monitoring over 24 h was normal. A brow recontouring operation was performed to improve cosmetic appearance.

Clinical appearance of subjects with craniosynostosis and FLNA variants. (a, b, c) Patient 1 aged 11 years, FMD with pansynostosis showing down-slanting palpebral fissures (a), irregular retroclined forehead with prominent supraorbital rim (b) and 3D CT reconstruction showing prominent supraorbital rim (c). (d–f) Patient 2 aged 7 years, OPD2 carrier with sagittal synostosis showing prominent supraorbital rim and hypertelorism (d, e) and broad halluces (f). (g–j) Patient 3, FMD with sagittal and bilambdoid synostosis showing down-slanting palpebral fissures at the age of 10 months (g, h), and spatulate fingertips and 2–3 toe syndactyly at the age of 1.5 years (i, j). (k–m) Patient 4 aged 3 years, FMD with prominent supraorbital rim and elongated, saddle-shaped skull contour (k, l). 3D CT reconstruction and venogram (apical view) showing fusion of the metopic and sagittal sutures associated with metopic and supraorbital ridging (m).
A diagnosis of FMD was suspected and genetic testing demonstrated a heterozygous variant in FLNA (NCBI ref. NM_001110556.1), c.3394G>T (p.(Gly1132Trp)), confirming this (Table 1). Genetic testing of both parents was normal, indicating a de novo mutation.
Patient 2
This female subject, the only child of healthy unrelated parents, was born 2 weeks post term via normal vaginal delivery. She presented perinatally with a cleft palate that was repaired at the age of 1 year. At the age of 7 years she was referred for craniofacial assessment because of hypertelorism, slight vertex prominence, and a prominent supraorbital ridge (Figure 1d and e). Clinical examination revealed broad thumbs and great toes (Figure 1f). 3D CT scanning demonstrated sagittal synostosis and decreased intracranial space; however, the ICP measured over 48 h was normal. She underwent calvarial expansion by lateral panel release with brow recontouring. Subsequently, her forehead prominence slowly progressed and she had a second procedure for further recontouring of her forehead aged 20 years. Currently, she maintains a full-time job as an administrator.
An initial clinical diagnosis of mild Pfeiffer syndrome was made. Genetic testing of FGFR2 was normal, whereas extended sequencing of FGFR1 (NCBI ref. NM_015850.3) revealed a heterozygous c.1408C>T (p.(Arg470Cys)) variant. The clinical significance of this finding remains unclear. Pfeiffer syndrome is uniquely caused by p.(Pro252Arg) mutations in FGFR1,12 and although a different heterozygous substitution at the same FGFR1 codon has been associated with hypogonadotrophic hypogonadism in the context of digenic inheritance,13 the patient had normal pubertal development and menstruation.
In view of the progressive frontal bossing and a need for a second recontouring operation, FLNA analysis was requested, which revealed a heterozygous c.760G>A (p.(Glu254Lys)) variant (Table 1). The identical mutation was previously described in four families segregating OPD2;5, 7, 8, 14 the subject’s mother was negative for the mutation and the father was not available for testing. The variant is likely to have arisen de novo, as all males previously described with this mutation have had lethal OPD2 phenotypes.7, 8, 14
Patient 3
This male subject, the only child of unrelated parents was born at 37+1 weeks’ gestation by emergency caesarean section for breech presentation. The pregnancy had been closely monitored due to an increased renal size noted at 32 weeks and oligohydramnios. He required a vesicostomy on the first day of life, secondary to urethral stenosis. From weaning he developed an aversion to oral feeding, resulting in a percutaneous endoscopic gastrostomy. He was treated in a Pavlik harness for developmental dysplasia of the hip.
On clinical genetic assessment at the age of 9 months, he was noted to be dysmorphic with deeply set, down-slanting eyes, disorganised eyebrows, a broad philtrum with a small mouth, a thin upper lip and a tongue tie (Figure 1g and h). He had poor muscle development in his shoulder girdle and widespread arthrogyroposis, involving large and small joints with marked ulnar deviation at the wrist and metacarpophalangeal joints. In addition, he had marked spatulate tips to all digits, with mild cutaneous syndactyly between the digits of his hands and partial 2, 3 toe syndactyly (Figure 1i and j). A skeletal survey was not considered diagnostic, although marked glenoid hypoplasia was noted.
Owing to clinical suspicion of an OPDS disorder, testing of FLNA was performed. This demonstrated hemizygosity for a previously undescribed variant c.3467C>T (p.(Pro1156Leu)), which was not present in the mother and had therefore arisen de novo (Table 1). A clinical diagnosis of FMD was proposed, given that the variant localises to the Repeat 9 domain close to the mutation hotspot for these cases.9
He was referred for urgent craniofacial assessment at the age of 18 months with a short history of vomiting and evidence of developmental delay. He was noted to have plagiocephaly with flattening of the left brow and reciprocal posterior occipital flattening on the right, without significant scaphocephaly. CT scanning showed sagittal synostosis but with posterior copper-beaten appearance and bone thinning; ICP monitoring over 24 h showed significantly raised ICP (mean=20 mm Hg, B-waves=8). He underwent subtotal calvarial remodelling at the age of 21 months; at operation it was noted that both lambdoid sutures were fused in addition to the sagittal suture. Following surgery, there was the impression of a marked behavioural change at the age of 24 months, with a reduction in temper tantrums and screaming, and he started to walk.
Patient 4
This male subject (Figure 1k and l), the only child of unrelated parents, was born at term by emergency caesarean section as the umbilical cord was around his wrist. The pregnancy was uneventful except that his mother took lamotrigine 75 mg daily for control of epilepsy and remained seizure-free. He was referred to the craniofacial clinic aged 14 months owing to concerns about his head shape. He had a history of coarse breathing attributed to laryngomalacia, an umbilical hernia, and fluctuating hearing loss associated with chronic otitis media with effusions. There was mild delay in expressive language and fine motor performance. On examination, he had scaphocephaly, a prominent supraorbital rim, mild exorbitism, and retrognathia. In addition, he had slightly broad flattened thumbs with some ulnar deviation and contractures around the wrists, elbows, knees, and ankles. 3D CT scanning showed sagittal synostosis and right frontotemporal and insular cortical thickening, suspicious of perisylvian polymicrogyria. There was no clinical or radiological evidence of raised ICP so this was not formally measured.
At the age of 15 months he underwent microlaryngoscopy and bronchoscopy, with insertion of bilateral grommets and adenoidectomy. He was found to have grade II congenital subglottic stenosis with a persistent thick anterior cricoid bar, for which he underwent laryngotracheal reconstruction aged 2.5 years. Unfortunately, he developed graft prolapse and required an emergency tracheostomy a month later that remained in situ until successful tracheal resection aged 3.5 years.
Repeat 3D CT scanning at 3 years of age (Figure 1m) demonstrated sagittal and metopic craniosynostosis and prominent metopic and supraorbital ridges. He underwent subtotal calvarial remodelling and burr remodelling to the glabella/metopic ridge with a good result. At follow-up he still had developmental delay and was awaiting further neurological assessment for possible absence seizures. A skeletal survey aged 3 years showed C2 pars defects, there was also mild undermodelling of the short tubular bones and the metaphyses of the long bones in the lower limbs, and mild tibial bowing.
In the family history, his mother had a prominent supraorbital ridge, which had been subject to a burring procedure aged 23 years; she had developed seizures from the age of 24 years. She was otherwise in good health with normal intellect. On review she had a broad, tall forehead, mild hypertelorism and exorbitism a long, triangular chin, and normal skull vault contour. She had anterior-posterior flattening of her chest with marked pectus excavatum, long fingers with camptodactyly of digits 2–5 and camptodactyly of toes 3–5, with lateral deviation of both halluces. There was no other family history of note.
An initial diagnosis of Shprintzen–Goldberg syndrome was suggested; however, sequencing of TGFBR1, TGFBR2, and SKI, as well as analysis for common mutations in FGFR2 and FGFR3, was normal. Subsequently, as the airway investigations and details of maternal history became available, a diagnosis of FMD was suspected. Genetic testing of FLNA showed a previously undescribed c.5169T>G (p.(Cys1723Trp)) substitution, present in hemizygous state in the patient and heterozygous state in his mother (Table 1). This variant lies in a region where other FMD mutations have been described,9 is not listed in large databases such as the Exome Aggregation Consortium (http://exac.broadinstitute.org/), and is predicted probably damaging (HumVar score 0.95) by Polyphen 2,15 supporting a causative role in the phenotype.
Discussion
We identified four unrelated patients, two females and two males, who have craniosynostosis associated with different missense substitutions in FLNA, only one of which has been reported previously. The encoded filamin A protein has an important role in membrane stability, crosslinking F-actin, and binding other proteins consistent with functions in both integration of cell signalling and cytoskeletal integrity. OPDS mutations cluster in several distinct regions and are thought to represent phenotypes associated with gain or altered function; biochemical evidence supporting this is available for the c.760G>A (p.(Glu254Lys)) mutation, present in patient 2.16
To our knowledge, there have been two published reports describing the association of craniosynostosis with FLNA mutations in single cases, one in a male with bicoronal synostosis and FMD,9 the other in a female with multisuture synostosis and MNS.10 Table 1 summarises the mutation analysis and clinical features of the four new patients, together with those previously published; patients are ordered according to the position of the substitution within the filamin A protein.
Scrutiny of Table 1 enables three broad conclusions. First, the six craniosynostosis-associated variants do not cluster in one part of the protein, but occur in three different regions (CHD2, Repeat 9, and Repeat 13/14 or 15), each of which contains two different variants; these correspond to three of the four regions of FLNA within which OPDS mutations tend to cluster.9 Interestingly, two of the three novel variants occurred in Repeat 9 in which only one mutation (c.3446C>T encoding p.(Pro1149Leu) in female with FMD)9 has previously been recorded. Second, FMD was the most frequently proposed clinical diagnosis (four of six, including all three affected males), but the manifesting carrier state for OPD2 and MNS were each present in a single affected female. Third, although there was significant variability in the combination of cranial sutures affected, in all but one case the sagittal suture was involved. In addition, in five out of six cases multiple sutures were affected (the one exception being Patient 2, the female OPD2 carrier with isolated sagittal synostosis). The overall impression is that craniosynostosis represents a low frequency association that might occur with a broad range of FLNA mutations conferring gain-of-function to the protein, and it is noteworthy that two of these were located in Repeat 9. The predisposition to craniosynostosis most likely reflects a generalised tendency for increased osteogenic differentiation in the cranial sutures.
In summary, our observations emphasise the importance of considering a filaminopathy in the differential diagnosis of craniosynostosis, especially when atypical clinical features are present, for example, laryngeal or urinary tract stenosis. The clinical presentation of OPDS is diverse and the craniofacial features and digital contractures become more prominent with age; in all four newly described patients, the onset of craniosynostosis was insidious, with the diagnosis not made until the second year of life in the two males, and later in childhood in the two females, so a high index of clinical suspicion is necessary. Craniofacial features that could trigger the search for a FLNA mutation include a prominent supraorbital rim, down-slanting palpebral fissures, and micrognathia or retrognathia. In addition, the presence of broad, flattened, or spatulate digits would strengthen the case for mutation analysis, especially when associated with camptodactyly or more generalised arthrogryposis. Apart from the importance for genetic counselling, early recognition of the diagnosis should alert the clinician to the possibility of raised ICP, which was documented in one of our four patients.
References
Eksioglu YZ, Scheffer IE, Cardenas P et al: Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron 1996; 16: 77–87.
Gargiulo A, Auricchio R, Barone MV et al: Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet 2007; 80: 751–758.
Bernstein JA, Bernstein D, Hehr U, Hudgins L : Familial cardiac valvulopathy due to filamin A mutation. Am J Med Genet A 2011; 155A: 2236–2241.
Reinstein E, Frentz S, Morgan T et al: Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in filamin A. Eur J Hum Genet 2013; 21: 494–502.
Robertson SP, Twigg SR, Sutherland-Smith AJ et al: Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet 2003; 33: 487–491.
Dudding BA, Gorlin RJ, Langer LO : The oto-palato-digital syndrome. A new symptom-complex consisting of deafness, dwarfism, cleft palate, characteristic facies, and a generalized bone dysplasia. Am J Dis Child 1967; 113: 214–221.
Young K, Barth CK, Moore C et al: Otopalatodigital syndrome type II associated with omphalocele: report of three cases. Am J Med Genet 1993; 45: 481–487.
Robertson S, Gunn T, Allen B, Chapman C, Becroft D : Are Melnick-Needles syndrome and oto-palato-digital syndrome type II allelic? Observations in a four-generation kindred. Am J Med Genet 1997; 71: 341–347.
Robertson SP, Jenkins ZA, Morgan T et al: Frontometaphyseal dysplasia: mutations in FLNA and phenotypic diversity. Am J Med Genet A 2006; 140: 1726–1736.
Foley C, Roberts K, Tchrakian N et al: Expansion of the spectrum of FLNA mutations associated with Melnick-Needles syndrome. Mol Syndromol 2010; 1: 121–126.
Sun Y, Almomani R, Aten E et al: Terminal osseous dysplasia is caused by a single recurrent mutation in the FLNA gene. Am J Hum Genet 2010; 87: 146–153.
Rossi M, Jones RL, Norbury G et al: The appearance of the feet in Pfeiffer syndrome caused by FGFR1 P252R mutation. Clin Dysmorphol 2003; 12: 269–274.
Pitteloud N, Quinton R, Pearce S et al: Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 2007; 117: 457–463.
Savarirayan R, Cormier-Daire V, Unger S et al: Otopalatodigital syndrome, type II: report of three cases with further delineation of the chondro-osseous morphology. Am J Med Genet 2000; 95: 193–200.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Clark AR, Sawyer GM, Robertson SP, Sutherland-Smith AJ : Skeletal dysplasias due to filamin A mutations result from a gain-of-function mechanism distinct from allelic neurological disorders. Hum Mol Genet 2009; 18: 4791–4800.
Acknowledgements
We thank Michael Oldridge for genetic analysis and the Wellcome Trust (102731 to AOMW) and Curekids NZ (SPR) for funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Fennell, N., Foulds, N., Johnson, D. et al. Association of mutations in FLNA with craniosynostosis. Eur J Hum Genet 23, 1684–1688 (2015). https://doi.org/10.1038/ejhg.2015.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.31
This article is cited by
-
Apert syndrome: magnetic resonance imaging (MRI) of associated intracranial anomalies
Child's Nervous System (2018)
-
Otopalatodigital spectrum disorders: refinement of the phenotypic and mutational spectrum
Journal of Human Genetics (2016)
