
Abstract
Acyl-CoA dehydrogenase family, member 9 (ACAD9) mutation is a frequent, usually fatal cause of early-onset cardiac hypertrophy and mitochondrial respiratory chain complex I deficiency in early childhood. We retrospectively studied a series of 20 unrelated children with cardiac hypertrophy and isolated complex I deficiency and identified compound heterozygosity for missense, splice site or frame shift ACAD9 variants in 8/20 patients (40%). Age at onset ranged from neonatal period to 9 years and 5/8 died in infancy. Heart transplantation was possible in 3/8. Two of them survived and one additional patient improved spontaneously. Importantly, the surviving patients later developed delayed-onset neurologic or muscular symptoms, namely cognitive impairment, seizures, muscle weakness and exercise intolerance. Other organ involvement included proximal tubulopathy, renal failure, secondary ovarian failure and optic atrophy. We conclude that ACAD9 mutation is the most frequent cause of cardiac hypertrophy and isolated complex I deficiency. Heart transplantation in children surviving neonatal period should be considered with caution, as delayed-onset muscle and brain involvement of various severity may occur, even if absent prior to transplantation.
Similar content being viewed by others


Introduction
The mitochondrial respiratory chain is composed of four multiprotein complexes (Complex I–IV), embedded in the inner mitochondrial membrane and ATP synthase (Complex V) allows ATP synthesis. Complex I is the largest mitochondrial respiratory chain complex and its deficiency accounts for almost one-third of respiratory chain disorders. Specific assembly factors, such as acyl-CoA dehydrogenase family, member 9 (ACAD9, NM_014049.4, mapping to 3q21.3), play an essential role in complex I assembly. It has been recently shown that ACAD9 variants account for severe, frequently lethal autosomal recessive cardiac hypertrophy in infancy.1, 2, 3, 4, 5 Studying a series of 20 unrelated children with early-onset cardiac hypertrophy and isolated complex I deficiency, we found ACAD9 variants in 8/20, three of whom received heart transplantation. We show here that ACAD9 variants are the prevalent cause of cardiac hypertrophy and isolated complex I deficiency in childhood. Heart transplantation in this frequent cause of cardiac hypertrophy should be considered with caution as patients surviving early-onset cardiac hypertrophy and recipients of heart transplantation developed delayed-onset multiorgan involvement.
Patients and methods
A total of 20 patients aged 0–9 years (11 males, 9 females) were included in this retrospective study over a period of 20 years at the Pediatric Cardiology Unit, Hôpital Necker-Enfants Malades, Paris. Inclusion criteria were (i) ultrasound evidence of cardiac hypertrophy (left ventricular wall thickness Z-score above +2), (ii) normal heart ultrasound in the parents, (iii) enzymological evidence of isolated complex I deficiency on frozen heart or skeletal muscle biopsy and (iv) exclusion of other known causes of cardiac hypertrophy, namely lysosomal storage disease, fatty acid oxidation disorders, Noonan syndrome and Friedreich ataxia.
Spectrophotometric assays of respiratory chain enzymes were carried out as previously described.6
Mitochondrial suspension of cultured skin fibroblasts was obtained after suspending 50 μl of frozen cells in mitochondria extraction medium (1 ml, 20 mm Tris-HCl (pH 7.2), 250 mm sucrose, 2 mm EGTA, 40 mm KCl, 1 mg/ml BSA) supplemented with 0.01% digitonin and 10% Percoll. After 10 min at 4 °C, the sample was centrifuged for 5 min at 5000 r.p.m. and the pellet was washed in extraction medium (1 ml) with 1 mg/ml BSA and subsequently centrifuged for 5 min at 5000 r.p.m. The cell pellet was suspended in 30 μl of extraction medium before enzyme measurement.
Exome sequencing was performed as previously reported7 and followed by Sanger sequencing. The exons and exon–intron boundaries of the ACAD9 gene (NM_014049.4) were amplified using specific primers1 after an initial denaturation at 96 °C for 5 min, followed by 30 cycles of 96 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, and a last extension at 72 °C for 10 min. Amplification products were purified by ExoSap IT (Amersham, Buckinghamshire, UK) and directly sequenced using the PRISM Ready Reaction Sequencing Kit (Perkin-Elmer, Oak Brook, IL, USA) on an automatic sequencer (ABI 3130xl; PE Applied Biosystems, Foster City, CA, USA). The ACAD9 variations have been submitted to ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/). Submission ID numbers are: Variation ID: 217744; variation ID: 217738; Variation ID: 217741; Variation ID: 217740; Variation ID: 217742; Variation ID: 217737; Variation ID: 217743; Variation ID: 217739. These variations are available at this url: http://www.ncbi.nlm.nih.gov/clinvar/submitters/505590/.
URL or websites
Alamut Visual: http://www.interactive-biosoftware.com/alamut-visual/; Polyphen-2: http://genetics.bwh.harvard.edu/pph2/; SIFT: http://sift.jcvi.org/; MutationTaster: http://www.mutationtaster.org/; ClinVar: http://www.ncbi.nlm.nih.gov/clinvar/.
Results
Whole-exome sequencing in 5/20 patients with cardiomyopathy and complex I deficiency detected compound heterozygosity for ACAD9 variants in 4/5 (patients 3–6) and homozygosity for an acylglycerol kinase (AGK, NM_018238.3) variant in one. This prompted us to sequence exons and exon–intron boundaries of the ACAD9 gene by Sanger sequencing in the remaining 15 patients and detect compound heterozygosity for missense or splice site ACAD9 variants in four additional patients (patients 1–2, 7–8). Therefore, the ACAD9 gene accounted for 8/20 cases of cardiomyopathy and complex I deficiency in our series (40%). In the remaining patients, the candidate gene approach identified nuclear DNA variants in 3/20 (NDUFS4 (NM_002495.2): 2/20 and C20ORF78 (NM_001242671.1): 1/20) and a mtDNA variant in 1/20 (ND5, NC_011137.1).
All but three ACAD9 variants were hitherto unreported. Novel mutations included one nonsense, two splice site, one initiation codon and seven missense variants (Table 1 and Supplementary Table S1). All amino acid changes but one (c.976G>C, previously reported2) were predicted to be deleterious using three different prediction softwares, namely SIFT, MutationTaster and Polyphen-2. Splice site variants were predicted to be deleterious as well by Alamut Visual. Parents were heterozygotes for the variants.
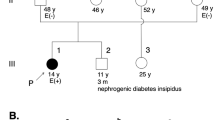
Heart failure or cardiovascular collapse was consistent presenting symptom in 8/8 probands and their affected relatives (5/5; Supplementary data). Age at onset ranged from the neonatal period to the first few months/years of life and heart ultrasounds consistently revealed severe cardiac hypertrophy (Z-score above 2). No overt extra-cardiac involvement was clinically present at the time of diagnosis in 6/8 children (Table 1), but all of them had elevated plasma lactate (4–20 mmol/l, normal below 2.4 mmol/l) and displayed mild to severe complex I deficiency in either heart or skeletal muscle (Table 1). All eight patients were given oral riboflavin for at least 2 months and none of them presented ultrasound evidence of responsiveness to the vitamin (not shown). None of the patients’ parents had cardiologic symptoms or ultrasound evidence of heart hypertrophy. The clinical presentation of the eight patients reported here is described in the Supplementary file section. No correlation between the type/location of the variant and the outcome of the disease could be found in our series.
Rapid progression of heart failure precluded organ transplantation in 4/8 children (patients 1–2 and 7–8), but 3/8 patients were proposed for heart transplantation as they had no overt extra-cardiac involvement at diagnosis (patients 3–5; Table 1). Patients 4 and 5 survived and did well during the following years. Importantly, the surviving patients (including the two living, transplanted cases 4–5) later developed delayed-onset neurologic or muscular symptoms, that is, cognitive impairment, seizures, muscle weakness and exercise intolerance. Other organ involvement included proximal tubulopathy, renal failure, secondary ovarian failure or optic atrophy (Table 1). Yet, while patients 5 and 6 enjoyed an almost normal personal and professional adult life, with employment, patient 4 experienced some learning difficulties and required special schooling.
Discussion
Here we report on compound heterozygosity for ACAD9 variants in 8/20 children with severe, early-onset cardiac hypertrophy with isolated complex I deficiency. Hence, ACAD9 variants should be regarded as the most frequent cause of isolated complex I deficiency in childhood cardiac hypertrophy. We suggest therefore giving priority to this gene when investigating patients with cardiac hypertrophy and isolated complex I deficiency in heart or skeletal muscle biopsy. Owing to the absence of extra-cardiac involvement at diagnosis, 3/8 patients benefited from heart transplantation in childhood or early adulthood. Two of them survived and one additional patient improved spontaneously. Long-term survey of their transplanted heart function is satisfactory. This is, to our knowledge, the first example of successful heart transplantation in ACAD9 mutations.
Yet, while lifespan of 2/3 transplanted patients was markedly improved, compared with the frequently fatal outcome of ACAD9 variants, long-term follow-up revealed late-onset multiorgan involvement, as commonly observed in the natural history of respiratory chain deficiency. Main clinical features included neurological and muscular symptoms, namely cognitive impairment, seizures, muscle weakness and exercise intolerance. Other organ involvement included proximal tubulopathy, renal failure, secondary ovarian failure and optic atrophy in our series.
Recent studies from several groups have previously reported ACAD9 variants in early-onset heart failure (6/15 deceased patients).1, 2, 3, 4 Importantly, surviving patients (9/15) experienced exercise intolerance and muscle weakness. Other features included cognitive impairment (1/15), stroke-like episodes (1/15) and liver failure (3/15; Table 2).
ACAD9 variation is not the unique example of delayed-onset multiorgan involvement in spontaneous survivors of respiratory chain deficiency or following organ transplantation. Indeed, deoxyguanosine kinase (DGUOK)-deficient patients suffering from life-threatening liver failure benefited of liver transplantation but frequently developed severe neurological involvement caused by their underlying mitochondrial disease.8 Similarly, most Pearson Marrow-Pancreas syndrome patients recovering from pancytopenia, watery diarrhea and exocrine pancreatic dysfunction further developed Kearns-Sayre syndrome.9 Recently, long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene variant has been reported in a 20-year-old woman who initially presented as a floppy infant but whose condition progressed during childhood and adolescence with increasing muscle weakness, focal motor seizures and optic atrophy.10
In conclusion, while a significant proportion of respiratory enzyme chain deficient patients may initially benefit from heart or liver transplantation, the decision to carry out organ transplantation remains difficult as neurological symptoms may later occur despite their absence prior to transplantation. To our opinion, however, the risk–benefit assessment seems in favor of organ transplantation, especially when a life-threatening symptom dominates the short-term prognosis, as it is the case in ACAD9 variants.
References
Gerards M, van den Bosch BJ, Danhauser K et al: Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain 2011; 134: 210–219.
Haack TB, Danhauser K, Haberberger B et al: Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet 2010; 42: 1131–1134.
Haack TB, Haberberger B, Frisch EM et al: Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet 2012; 49: 277–283.
Nouws J, Nijtmans L, Houten SM et al: Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab 2010; 12: 283–294.
Scholte HR, Busch HF, Bakker HD, Bogaard JM, Luyt-Houwen IE, Kuyt LP : Riboflavin-responsive complex I deficiency. Biochim Biophys Acta 1995; 1271: 75–83.
Rustin P, Chretien D, Bourgeron T et al: Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 1994; 228: 35–51.
Haack TB, Hogarth P, Kruer MC et al: Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012; 91: 1144–1149.
Grabhorn E, Tsiakas K, Herden U et al: Long-term outcomes after liver transplantation for deoxyguanosine kinase deficiency: a single-center experience and a review of the literature. Liver Transplant 2014; 20: 464–472.
Simonsz HJ, Barlocher K, Rotig A. : Kearns-Sayre's syndrome developing in a boy who survived pearson's syndrome caused by mitochondrial DNA deletion. Doc Ophthalmol 1992; 82: 73–79.
Tuppen HA, Fehmi J, Czermin B et al: Long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene mutation. Mol Genet Metab 2010; 100: 345–348.
Calvo SE, Compton AG, Hershman SG et al: Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012; 4: 1–15.
Acknowledgements
The authors thank the families for their participation. This work was supported in part by Association Française contre les Myopathies (AFM). MDM is supported by a fellowship from the AFM (16615), by German Bundesministerium für Bildung und Forschung (BMBF) through the German Network for mitochondrial disorders (mitoNET, 01GM1113C to HP) and through the E-Rare project GENOMIT (01GM1207).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Collet, M., Assouline, Z., Bonnet, D. et al. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur J Hum Genet 24, 1112–1116 (2016). https://doi.org/10.1038/ejhg.2015.264
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.264
This article is cited by
-
Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective?
Orphanet Journal of Rare Diseases (2018)
-
Mitochondrial Fatty Acid Oxidation Disorders Associated with Cardiac Disease
Current Pathobiology Reports (2017)
