
Abstract
Alopecia with mental retardation (APMR) is a very rare disorder. In this study, we report on a consanguineous Pakistani family (AP91) with mild-to-moderate intellectual disability, adolescent alopecia and dentogingival abnormalities. Using homozygosity mapping, linkage analysis and exome sequencing, we identified a novel rare missense variant c.898G>A (p.(Glu300Lys)) in ITGB6, which co-segregates with the phenotype within the family and is predicted to be deleterious. Structural modeling shows that Glu300 lies in the β-propeller domain, and is surrounded by several residues that are important for heterodimerization with α integrin. Previous studies showed that ITGB6 variants can cause amelogenesis imperfecta in humans, but patients from family AP91 who are homozygous for the c.898G>A variant present with neurological and dermatological features, indicating a role for ITGB6 beyond enamel formation. Our study demonstrates that a rare deleterious variant within ITGB6 causes not only dentogingival anomalies but also intellectual disability and alopecia.
Similar content being viewed by others
Introduction
Although the nervous system, tooth enamel, mucosal membranes and epidermis are ectodermal in origin, human diseases that affect all four ectodermal derivatives are rare. A few genes have been associated with phenotypes including alopecia and mental retardation (APMR), namely: DCAF17 (2q31.1, MIM 612515) for Woodhouse–Sakati syndrome (MIM 241080), which includes endocrine disease and hearing impairment;1 ALX4 (11p11.2, MIM 605420) for frontonasal dysplasia 2 (MIM 613451) with APMR;2 and MBTPS2 (Xp22.12-p22.11, MIM 300294) for ichthyosis follicularis, atrichia and photophobia with or without intellectual disability or Bresheck syndrome (MIM 308205).3 Four autosomal recessive APMR loci have been mapped, namely APMR1 on 3q26.33-q27.3,4 APMR2 on 3q26.2-q26.31,5 APMR3 on 18q11.2-q12.2(ref. 6) and APMR(4) on 1p31.1-p22.3,7 but the causal variants within these loci have not been identified. Here we report on a variant in ITGB6 (MIM 147558), which encodes integrin beta-6, as the cause of autosomal recessive APMR with premature skin aging and dentogingival abnormalities.
Materials and methods
The study was approved by the Institutional Review Boards of Quaid-i-Azam University and Baylor College of Medicine and Affiliated Hospitals. Informed consent was obtained from participating family members, including permission to publish facial photographs. For family AP91 (Figure 1a), homozygosity mapping was carried out using genome-wide genotypes for ~400 microsatellite markers. Parametric multipoint linkage analysis was performed with MERLIN8 using autosomal recessive inheritance with complete penetrance. A DNA sample from affected individual IV-6 was submitted for exome sequencing at the University of Washington Center for Mendelian Genomics. Pathogenicity of identified variants was evaluated using multiple bioinformatics tools (Table 1). For each variant, minor allele frequencies (MAF) were checked in variant databases 1000 Genomes, Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) and in 148 in-house exomes from unrelated Pakistani individuals with Mendelian traits that do not overlap with the phenotypes in family AP91. Because of the rarity of the phenotype in family AP91, variants that were homozygous in the in-house exomes were excluded. Rare potentially pathogenic variants were Sanger-sequenced in the rest of family AP91 and the ITGB6 variant was sequenced in 248 Pakistani control individuals. The ITGB6 variant was submitted to ClinVar. To check for evolutionary conservation of ITGB6 amino acid residues, the human sequence was aligned with 51 vertebrate sequences. Phyre2(ref. 9) was used for molecular modeling. RT-PCR was performed on commercially available human cDNA (Clontech, Mountain View, CA, USA).

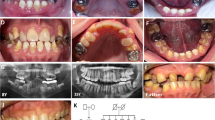
Pedigree drawing, clinical information and chromatograms from family AP91. (a) Pedigree of consanguineous family AP91 with intellectual disability, alopecia, progeroid appearance, periodontal disease and tooth abnormalities co-segregating with the ITGB6 c.898G>A (p.(Glu300Lys)) variant. (b) Photographs of affected individuals IV-2, IV-3 and IV-6 at 40, 39 and 33 years of age, respectively. Note fine sparse scalp hair, sparse eyebrows and eyelashes and deeply set facial wrinkles. Women were veiled because of religious beliefs. Affected male IV-2 had bilateral super-eruption of maxillary lateral incisors, small central incisors and poor alignment of teeth. The affected youngest sister IV-6 also had misaligned mandibular anterior teeth with rough surfaces. Both IV-2 and IV-6 had prominent yellowish-brown tooth stains and severe gingival disease. Individual IV-3 had no remaining teeth. (c) Chromatograms from homozygous affected and heterozygous/wildtype unaffected family members.
Results
Family AP91 is a four-generation consanguineous pedigree (Figure 1a) from the Galiyat region of Pakistan. Affected individuals had normal thickness of scalp hair at birth although hair was easy to pluck. Progressive hair loss and thinning started at ages 10–12 years. Affected male individual IV-2 (Figure 1b) had obvious scalp hair thinning and lacked moustache and beard hair. All three affected individuals had sparse eyebrows and eyelashes, and early-onset skin aging with deeply set facial wrinkles, giving them a progeroid appearance. Since childhood, the affected individuals had poor speech and memory, were unable to go to school, wandered aimlessly in public places, exhibited aggressive behavior and/or lacked the concept of danger or fear. Based on the Wechsler Adult Intelligence Scale test that was translated to the local dialect, the affected individuals had mild-to-moderate intellectual disability (IQ 53–61). They exhibited dental abnormalities such as plaques, calculi and gingival recession, which may partly be due to poor hygiene but can also be included in the disease spectrum. Individual IV-3 lost all her teeth, while individuals IV-2 and IV-6 had yellowish-brown tooth stains (Figure 1b). The lateral maxillary incisors of individual IV-2 were super-erupted and protruded anteriorly. For individual IV-6, alignment of maxillary teeth was normal, but the mandibular incisors and canines were badly aligned and had rough enamel surfaces. No additional syndromic features such as hearing, visual, cardiopulmonary or endocrine abnormalities were detected.
Both linkage interval and homozygous region identified in family AP91 were flanked by markers D2S1353 and D2S2307, defining a 15.89-Mb locus on chromosome 2q24.1-q31.1. Linkage analysis yielded a maximum multipoint LOD score of 2.65 (θ =0) for several markers in the mapped region. Within the homozygous region, exome sequencing revealed 80 homozygous variants, but only one variant, ITGB6 NM_000888.3:c.898G>A (p.(Glu300Lys)), did not occur in variant databases 1000 Genomes, EVS or dbSNP, but was heterozygous in the ExAC database with an overall MAF of 3.3 × 10−5. The ITGB6 variant and three additional rare homozygous exome variants outside the mapped region (Table 1) did not occur in 148 in-house exomes. However Sanger sequencing using DNA samples from the rest of family AP91 revealed co-segregation of only the ITGB6 (Figures 1a and c) variant with the phenotypes. Additionally the ITGB6 variant was absent in 496 Pakistani control chromosomes, and is the only rare variant that not only lies within the mapped region and segregates with disease, but also occurs at a highly conserved residue and is predicted to be deleterious by the majority of bioinformatics results (Table 1).
The glutamic acid at position 300 of human ITGB6 is identical across all species and lies within a highly conserved region from His275 to Ser304 of human ITGB6 (Figure 2a). The Glu300 residue occurs at the predicted β-propeller domain and lies very close to several amino acids bearing key residues that are essential for the interface between α and β subunits of the integrin heterodimer10 (Figure 2a). Conversion of the negatively charged side chain of glutamic acid to the positively charged side chain of lysine is predicted to result in a more buried conformation and slight torsion of the hairpin loop between α-helices 4 and 5 (Figure 2b). Thus, it is predicted that the substitution p.Glu300Lys would affect proper contact at the predicted βA domain-propeller interface.10 Lastly RT-PCR revealed ITGB6 expression in adult but not fetal brain (Figure 2c).

Protein sequence alignment, molecular modeling and ITGB6 expression. (a) The glutamic acid at position 300 (E300) of the human sequence is identical across 51 vertebrate species, including 36 mammals, five birds, three reptiles, one amphibian and six fish, and lies within a highly conserved region that includes His275 to Ser304 of human ITGB6. (b) Molecular modeling predicts a more buried configuration due to the p.Glu300Lys change and slight torsional adjustments on the hairpin loop within the β-propeller domain. (c) RT-PCR of commercially obtained human cDNA shows ITGB6 expression in adult brain but not in fetal brain. Control denotes no template. Forward 5′-GAGGGGAAAACCATCATTCA-3′ and reverse 5′-GTAGGACAACCCCGATGAGA-3′ primers for ITGB6 exon 3 were used. ClinVar accession number for the ITGB6 variant is SCV000249619.
Discussion
ITGB6 variants were previously shown to cause amelogenesis imperfecta (AI) in young children,11, 12 and Itgb6-knockout mice present with enamel defects,13 periodontal disease14 and juvenile baldness.15 The occurrence of AI in knockout mice and in children <10 years old was supported by enamel defects seen by electron microscopy.11, 12, 13 Likewise the three adult affected individuals from family AP91 had tooth loss, yellowish-brown staining, malocclusion, poor alignment and/or rough enamel, all of which may be AI-related. In addition the affected AP91 family members had severe periodontal disease, which was also observed in Itgb6−/− mice.14 Hair loss due to ITGB6 variants has not been reported in humans, which may be due to the young age of previously ascertained probands.11, 12 Adolescent alopecia was observed in affected individuals from family AP91, consistent with juvenile baldness on the head and neck of Itgb6−/− mice.15
Although a previously reported AI child who is homozygous for a stop-gain ITGB6 variant seemed to have average intelligence despite being suspected of having Nance-Horan syndrome11 (MIM 302350), all three adult affected individuals in this report were confirmed to have mild-to-moderate intellectual disability by formal testing. Our RT-PCR result showed ITGB6 expression in adult but not fetal brain, suggesting a role for ITGB6 in maintenance of brain function.
Although Itgb6-knockout mice developed gradual, age-related emphysema in adulthood,16 lung problems were not reported in affected AP91 family members. The two older affected individuals died recently of severe diarrhea. For the living affected female IV-6, it is unknown whether she will eventually develop lung disease due to the ITGB6 variant.
In response to epithelial injury, ITGB6 activates latent transforming growth factor beta-1 (TGFβ-1) by binding to the arginine-glycine-aspartate motif of its latency-associated peptide, then the activated TGFβ-1 enhances healing or fibrosis and downregulates the inflammatory response.17 Lack of ITGB6, therefore, results in enhanced inflammatory response. In knockout mice, degenerating hair follicles were surrounded by foci of mononuclear cells15 and inflammatory infiltrates were enhanced in gingiva and lung,14, 16 but not in other epithelial tissues.17 While TGFβ-1 downregulation facilitates faster re-epithelialization post injury and protects against fibrosis,18 chronic lack of TGFβ-1 leads to failure of tissue regeneration resulting in epidermal hypoproliferation, blocked hair follicle growth and neurodegeneration in brain.19, 20 The phenotypes observed in family AP91 and Itgb6−/− mice are therefore consistent with failure of TGFβ-1 activation from ITGB6 deficiency.
In summary, we present ITGB6 as a gene that is involved in the pathophysiology of APMR, and an ITGB6 variant that causes a rare disease spectrum including not only dental anomalies but also intellectual disability, periodontal disease and adolescent alopecia, further lending support to the role of TGFβ-1 in regeneration and maintenance of ectodermal derivatives. Although children with ITGB6 variants were reported to have AI only, our findings suggest that they should be followed up over time and examined for additional phenotypes. This study also demonstrates the importance of careful follow-up phenotyping in families and individuals with congenital disease for other disorders that manifest at a later age.
References
Alazami AM, Al-Saif A, Al-Semari A et al: Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet 2008; 83: 684–691.
Kayserili H, Uz E, Niessen C et al: ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Genet 2009; 18: 4357–4366.
Oeffner F, Fischer G, Happle R et al: IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet 2009; 84: 459–467.
John P, Ali G, Chishti MS, Naqvi SM, Leal SM, Ahmad W : Localization of a novel locus for alopecia with mental retardation syndrome to chromosome 3q26.33-q27.3. Hum Genet 2006; 118: 665–667.
Wali A, John P, Gul A et al: A novel locus for alopecia with mental retardation syndrome (APMR2) maps to chromosome 3q26.2-q26.31. Clin Genet 2006; 70: 233–239.
Wali A, Ali G, John P et al: Mapping of a gene for alopecia with mental retardation syndrome (APMR3) on chromosome 18q11.2-q12.2. Ann Hum Genet 2007; 71: 570–577.
Tzschach A, Bozorgmehr B, Hadavi V et al: Alopecia-mental retardation syndrome: clinical and molecular characterization of four patients. Br J Dermatol 2008; 159: 748–751.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin – rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Kelley LA, Sternberg MJ : Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 2009; 4: 363–371.
Xiong JP, Stehle T, Diefenbach B et al: Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 2001; 294: 339–345.
Wang SK, Choi M, Richardson AS et al: ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum Mol Genet 2014; 23: 2157–2163.
Poulter JA, Brookes SJ, Shore RC et al: A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta. Hum Mol Genet 2014; 23: 2189–2197.
Mohazab L, Koivisto L, Jiang G et al: Critical role for αvβ6 integrin in enamel biomineralization. J Cell Sci 2013; 126: 732–744.
Ghannad F, Nica D, Fulle MI et al: Absence of alphavbeta6 integrin is linked to initiation and progression of periodontal disease. Am J Pathol 2008; 172: 1271–1286.
Huang XZ, Wu JF, Cass D et al: Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 1996; 133: 921–928.
Morris DG, Huang X, Kaminski N et al: Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003; 422: 169–173.
Munger JS, Huang X, Kawakatsu H et al: The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96: 319–328.
Puthawala K, Hadjiangelis N, Jacoby SC et al: Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med 2008; 177: 82–90.
Liu X, Alexander V, Vijayachandra K, Bhogte E, Diamond I, Glick A : Conditional epidermal expression of TGFbeta 1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia. Proc Natl Acad Sci USA 2001; 98: 9139–9144.
Buckwalter MS, Wyss-Coray T : Modelling neuroinflammatory phenotypes in vivo. J Neuroinflammation 2004; 1: 10.
Acknowledgements
We thank the family members for their invaluable cooperation and participation in this study. This work was supported by National Institutes of Health grant U54 HG006493 and by the Higher Education Commission (HEC), Islamabad, Pakistan.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
University of Washington Center for Mendelian Genomics (UWCMG) Michael J Bamshad1,2, Jay Shendure1, Deborah A Nickerson1, Gonçalo R Abecasis3, Peter Anderson1, Marcus Annable1, Mallory Beightol1, Brian L Browning1, Kati J Buckingham1, Christina Chen1, Jennifer Chin1, Jessica X Chong1, Gregory M Cooper4, Colleen Davis1, Lindsay Felker1, Christopher Frazar1, David Hanna1, Zongxiao He5, Preti Jain4, Gail P Jarvik1, Eric Johanson1, Goo Jun3, Martin Kircher1, Tom Kolar1, Suzanne M Leal5, Daniel Luksic1, Margaret J McMillin1, Sean McGee1, Brenton Munson1, Brian J O'Roak1, Bryan Paeper1, Karynne Patterson1, Eric Phillips1, Jessica Pijoan1, Christa Poel1, Peggy D Robertson1, Regie Lyn P Santos-Cortez5, Tristan Shaffer1, Cindy Shephard1, Deborah L Siegel1, Joshua D Smith1, Jeffrey C Staples1, Holly K Tabor1,2, Monica Tackett1, Gao T Wang5 and Qian Yi11University of Washington, Seattle, WA, USA; 2Seattle Children’s Hospital, Seattle, WA, USA; 3University of Michigan, Ann Arbor, MI, USA; 4HudsonAlpha Institute of Technology, Huntsville, AL, USA; 5Baylor College of Medicine, Houston, TX, USA
Rights and permissions
About this article

Cite this article
Ansar, M., Jan, A., Santos-Cortez, R. et al. Expansion of the spectrum of ITGB6-related disorders to adolescent alopecia, dentogingival abnormalities and intellectual disability. Eur J Hum Genet 24, 1223–1227 (2016). https://doi.org/10.1038/ejhg.2015.260
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.260
