
Abstract
A rare genetic variant in the desmosomal gene plakophilin-2 (PKP2) c.419C>T(p.(S140F)) has repeatedly been identified in patients with dilated cardiomyopathy (DCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC). Whether this is a disease-causing variant remains highly controversial. We tested this hypothesis using three approaches. Initially, in a prospective study of 10 407 individuals from the general population, including 2688 who developed heart failure or arrhythmias during >14 years of follow-up, PKP2 c.419C>T was identified in 98 individuals (0.94%). PKP2 genotype was not associated with electrocardiographic or echocardiographic changes, or with plasma levels of probrain natriuretic peptide (all P≥0.05). In c.419C>T carriers versus non-carriers, multifactorially adjusted hazard ratios were 1.26 (95% confidence interval: 0.77–2.07) for heart failure, 1.40 (0.90–2.17) for arrhythmias, 1.15 (0.78–1.71) for end points combined, and 1.33 (0.98–1.80) for all-cause mortality. The cumulative survival as a function of age and PKP2 genotype was similar among carriers and non-carriers (P=0.14). Second, comparing 517 patients referred for genetic testing with 1918 matched controls, odds ratios as a function of c.419C>T genotype were 2.11 (0.50–8.99) for ARVC, 0.72 (0.16–3.28) for hypertrophic cardiomyopathy (HCM)/DCM, and 1.28 (0.46–3.54) for end points combined. Third, in in vitro studies cellular localization of plakophilin-2, plakoglobin, connexin-43, or N-cadherin were similar in cells transfected with wild-type or mutant plakophilin-2. In conclusion, combining epidemiological data, with data on patients referred for genetic testing for ARVC or HCM/DCM, and data from in vitro studies, PKP2 c.419C>T did not associate with heart failure, arrhythmias, or premature death, with ARVC or HCM/DCM, or with effects in vitro, suggesting that this is not a disease-causing variant.
Similar content being viewed by others


Introduction
Cardiomyopathies represent a heterogenous group of heart muscle disorders characterized by variable degrees of systolic and/or diastolic dysfunction, chamber and wall dimension abnormalities, and diverse microscopic alterations. Major types of cardiomyopathy include hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic right ventricular cardiomyopathy (ARVC). Clinically these cardiomyopathies are associated with significant morbidity and mortality and with risk of sudden cardiac death.1
Genetic variation in numerous genes has been associated with cardiomyopathy. Mutations in desmosomal genes are a major cause of ARVC and may account for up to 50% of cases.2, 3 Desmosomal mutations have also been identified in DCM,4, 5 suggesting a close genetic link between these two clinical entities. Furthermore, several publications have reported a high percentage of ARVC probands carrying more than one desmosomal mutation,2, 6, 7 suggesting either that a single genetic ‘hit’ may not be sufficient to cause the full-blown phenotype or that some genetic variants are not uncommon in the general population, and therefore occur by chance together with disease-causing mutations. Associations are often based on cosegregation analyses in small families, absence in limited sized control populations, and/or inference of functionality in silico. Therefore, there is an urgent need to determine the genetic variants in desmosomal genes in large epidemiological studies of the general population, and the association of such variants with intermediate phenotypes and with prospective risk of relevant disease end points. As a supplement to the human data, selected variants may be characterized in in vitro cell studies.
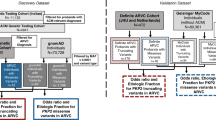
Plakophilin-2 (PKP2) c.419C>T (p.(S140F); rs150821281) has repeatedly been identified in patients with ARVC and DCM3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 (Supplementary Table 1). However, this association is highly controversial,8, 14 because both large prospective epidemiological data and in vitro data on this variant are lacking. Therefore, combining epidemiological data with patient data and in vitro studies, we tested the hypothesis that PKP2 c.419C>T associates with changes in the electrocardiogram (ECG) or echocardiogram and with risk of heart failure, arrhythmias, or premature death in >10 000 individuals from the general population. We also compared the odds ratios for ARVC, HCM/DCM, and long QT syndrome as a function of c.419C>T genotype in >500 patients referred for genetic testing with 1918 matched controls from the general population. Finally, we determined the effects of this variant in vitro. This is clinically important, as genetic testing is included in the updated ARVC diagnostic criteria,15 and therefore could have profound impact for both the proband and their families.
Materials and Methods
Participants
Prospective study
The Copenhagen City Heart Study (CCHS)16 is a prospective study of the Danish general population initiated in 1976–1978 with follow-up examinations in 1981–1983, 1991–1994, and 2001–2003. Participants were selected based on the National Danish Civil Registration System to reflect the adult Danish population aged 20–100+ years. Data were obtained from a self-administered questionnaire reviewed together with an investigator on the day of attendance, a physical examination, including an ECG, and from blood samples. In the present analyses, we included 10 407 participants from the 1991–1994 and 2001–2003 examinations for whom DNA was available. Of these, 1534 developed a heart failure end point, and 1902 developed arrhythmias during follow-up. Follow-up time began at the time of blood sampling and ended at the occurrence of event, emigration, or 10 May 2011 (last update of the registry), whichever came first.
Information on clinical events was drawn from the National Danish Patient Registry and the National Danish Causes of Death Registry from 1 January 1977 (the establishment of the National Danish Patient Registry) to 10 May 2011 (last update of the registry). The National Danish Patient Registry has information on all patient contacts with all clinical hospital departments and outpatient clinics in Denmark, including emergency wards (from 1994). The National Danish Causes of Death Registry contains data on the causes of all deaths in Denmark, as reported by hospitals and general practitioners.
All participants were white and of Danish descent. The study was approved by institutional review boards and by Danish ethical committees and conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants.
ECGs and echocardiograms
Standard 12 lead ECGs were recorded at rest in the supine position in all participants using a MAC 1200 electrocardiograph (GE Marquette Inc., Milwaukee, WI, USA) (1991–1994 examination) or a MAC-PC electrocardiograph (GE Marquette) (2001–2004 examination). ECGs were evaluated using the Minnesota Code system17 by two independent technicians, and in case of disagreement by a cardiologist. The following ECG definitions were used in the present study: sinus rhythm at baseline (absence of any Minnesota category 8 findings); complete or incomplete right bundle branch block (abnormality in category 7-2 or 7-3, respectively); any ventricular conductions defect (any finding in category 7); and left or right ventricular hypertrophy (any finding in category 3). Automated measurements of ECG intervals were available in 5485 participants, and analyzed using the Cardfile v.1.21 software (PPG Hellige GmbH, Freiburg, Germany).
Echocardiography was performed in 3453 participants using a Vivid Five Ultrasound machine with a 2.5 MHz probe (GE Vingmed, Horten, Norway). All subjects were scanned using a standardized protocol including standard apical and parasternal projections, 2-D and M-mode scans, and application of the standard 16-segment model. If image quality did not allow obtainment of M-mode images, only 2-D measurements were used. If both measures were available, the average was used. Left ventricular mass index was calculated as described previously.18
Clinical end points
We defined a heart failure end point as International Classification of Disease (ICD) codes for heart failure/cardiomyopathy (ICD8: 425.99, 427.09–11, 427.19, 427.99, 428.99, 429.08, 519.19, 782.49; ICD10: I42–43, I50, I51.5), dyspnea/edema (ICD8: 783.21, 783.29; ICD10: R06, R60), or myocarditis (ICD8: 74.29, 93.92, 391.29, 422.99; ICD10: I40–41, I51.4); and an arrhythmia end point as ICD codes for tachyarrhythmia (ICD8: 427.90–97, 782.19, 782.29; ICD10: I47–49, R00), syncope (ICD8: 782.59, ICD10: R55), or cardiac arrest (ICD10: I46). If more than one clinical end point was identified in the same participant, the first registered diagnosis was used in the analyses. For the baseline characteristics, presence of ischemic heart disease was defined as occurrence of relevant diagnostic codes (ICD8: 410–14; ICD10: I20–25). A total of 320 individuals (one PKP2 c.419C>T carrier and 319 non-carriers) experienced an event before study inclusion, and were excluded from further analyses.
Laboratory analyses
Plasma total cholesterol was measured using a standard colorimetric assay (Boehringer Mannheim, Mannheim, Germany). Plasma probrain natriuretic peptide (pro-BNP) was measured in a subset of participants (n=5813) from the 2001 to 2003 examination of the CCHS, as described previously.19
Other covariates
Body mass index was measured weight (in kilograms) divided by measured height in meters squared (kg/m2). Alcohol consumption was self-reported intake of units of alcohol consumed per week (one unit of alcohol=12 g). Smoking was active smoking. Hypertension was defined as a systolic blood pressure ≥140 mm Hg, a diastolic blood pressure ≥90 mm Hg, or use of antihypertensive therapy. Diabetes mellitus was self-reported disease, use of antidiabetic medication, or a non-fasting plasma glucose >11 mmol/l (198 mg/dl).
Case–control study
In a case–control study, we included 517 consecutive, unrelated patients referred for genetic testing to the Department of Clinical Biochemistry, Rigshospitalet, Copenhagen University Hospital. The referral diagnoses were ARVC (n=114), HCM or DCM (n=316), and long QT syndrome (n=87). Patients were matched 1:4 with controls (n=1918) from the CCHS without heart failure or arrhythmia end points as defined above. Matching was by sex and by age in the following age groups: 20–35 years and above 35 years in 5-year intervals. The study was in agreement with local ethics regulations.
Genotyping
An ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Carlsbad, CA, USA) and Taqman based assays were used to genotype participants in the CCHS and patients referred for genetic testing for PKP2 c.419C>T (p.(S140F); NM_004572.3; rs150821281), with the exception of patients referred for ARVC in whom genotyping was by direct sequencing of PKP2. Results were submitted to www.arvcdatabase.info.
In vitro analyses
For generation of cDNA constructs, full-length PKP2 cDNA was amplified from human heart total mRNA and cloned into the pGEM-T-easy vector (Promega, Madison, WI, USA). PKP2 cDNA was subsequently cloned in frame with green fluorescent protein (GFP) into the pEGFPN3 vector (Clontech, Saint-Germain-en-Laye, France) to obtain C terminally GFP-tagged PKP2 (pEGFPN3-PKP2). The PKP2 c.419C>T variant was inserted by direct-site mutagenesis (Agilent Technologies, Les Ulis, France). Primers are listed in Supplementary Table 2.
Neonatal rat cardiomyocytes were isolated as described previously20 with slight modifications. After isolation, cardiomyocytes were plated in a 24-well plate (300 000 cells per well) and cultured in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin/streptomycin. After 18 h of culture, 0.8 μg of wild-type or mutant pEGFPN3-PKP2 were transiently transfected using 2 μl of Lipofectamine 2000 (Life Technologies, Saint Aubin, France) in the Opti-MEM medium. After 4 h, the medium was replaced with 5% FBS-DMEM. After 4 days of culture, cells were immunolabelled. Cells were fixed for 5 min in 20 °C methanol, permeabilized by 0.1% Triton X-100-phosphate-buffered saline (PBS), and blocked for 1 h at room temperature with 5% bovine serum albumin-PBS. Cells were colabelled with GFP rabbit antibody (diluted 1:300, clone TP401; Clinisciences, Nanterre, France) or GFP mouse antibody (diluted 1:200; Roche, Basel, Switzerland), mouse anti-plakoglobin (diluted 1:200, clone 15F11; Sigma-Aldrich, Dorset, UK), mouse anti-N-cadherin (diluted 1:50, clone 3B9; Zymed, South San Francisco, CA, USA), or rabbit anti-connexin-43 (diluted 1:100; Zymed) antibodies. The slides were washed in PBS, and labeled with Alexa Fluor secondary antibodies (Invitrogen, Carlsbad, CA, USA). Pictures were analyzed with an Olympus IX50 fluorescent microscope (Olympus Corporation, Tokyo, Japan) and 3D deconvolution using the Metamorph software (Roper Scientific, Trenton, NJ, USA).
For proliferation experiments, HEK293T cells were plated in a 96-well plate (15 000 cells per well) and transfected with wild-type, mutant pEGFPN3-PKP2, or empty pEGFPN3 using JetPei (PolyPlus Transfection, Illkirch, France). After 24 h of culture, the bromodeoxyuridine (BrdU) incorporation was assessed using the ‘Cell Proliferation Elisa BrdU Colorimetric Assay’ as recommended by the manufacturer (Roche). Measurements were performed in triplicate and tests were repeated three times.
Statistical analyses
Data were analyzed by AHC, PRK, and AT-H using STATA SE version 12.1. χ2 Test was used to evaluate the Hardy–Weinberg equilibrium. Student’s t-test, Fisher’s exact test, Pearson’s χ2 test, and Kruskal-Wallis test of variance were used in two-group comparisons.
In the CCHS, Cox proportional hazard regression models with age as time-scale and delayed entry (left truncation) were used to estimate hazard ratios (HRs) for heart failure, arrhythmia end points, and total mortality as a function of genotype. Proportionality of hazards over time was judged by visual inspection of cumulative hazard logarithm plots against age; no violations were observed. Models were multifactorially adjusted for age, sex, total cholesterol, body mass index, alcohol consumption, smoking, hypertension, diabetes, and presence of ischemic heart disease at baseline. Individuals with events before inclusion in the study were excluded from the analyses.
Kaplan–Meier curves and log-rank test compared the probability of survival as a function of age and c.419C>T genotype. Cox proportional hazard models were used to estimate HR for all-cause mortality by PKP2 genotype. Models were multifactorially adjusted as described above.
In the case–control study comparing referred patients with event free controls from the CCHS, conditional logistic regression analyses estimated odds ratios for ARVC, HCM/DCM, long QT syndrome, or these diagnoses combined as a function of c.419C>T genotype.
Results
Prospective study
The CCHS
Baseline characteristics of the 10 407 participants in the CCHS as a function of PKP2 c.419C>T genotype are shown in Table 1. Baseline characteristics did not differ by genotype (P-values: 0.36–0.99).
Genotyping identified 97 (0.94%) heterozygotes, and one homozygote (0.01%) for c.419C>T in this general population study, corresponding to a minor allele frequency of ~0.5%. Genotype frequencies did not deviate from those predicted by the Hardy–Weinberg equilibrium (P=0.63). For practical purposes, heterozygotes and homozygote have been combined as ‘carriers’ throughout the study.
ECGs, echocardiograms and measures of heart failure
Paraclinical characteristics of participants in the CCHS as a function of PKP2 c.419C>T genotype are shown in Table 2. Neither Minnesota coding for resting heart rate, sinus rhythm at baseline, ventricular conduction defects, or hypertrophy, nor digitalized measurements of duration of P waves, PQ intervals, and QRS complexes differed between carriers and non-carriers (P-values: 0.15 to 0.98), although the QTc intervals tended to be shorter in carriers than in non-carriers (P=0.05).
Echocardiographic evaluation of left ventricular systolic and diastolic diameter, ejection fraction, and mass index also did not differ by c.419C>T genotype. In particular, all c.419C>T carriers had preserved left ventricular ejection fraction, as well as similar levels of pro-BNP compared with non-carriers.
Clinical end points
To achieve sufficient statistical power, the clinical events were divided into two groups: Group 1 heart failure end point, total n=1534: including heart failure/cardiomyopathy, dyspnea/edema and myocarditis; Group 2 arrhythmia end point, total n=1902: including tachyarrhythmia, syncope, and cardiac arrest; or groups 1 and 2 combined, n=2688. The multifactorially adjusted HRs were 1.26 (95% confidence interval (CI): 0.77–2.07) for heart failure end points, 1.40 (95% CI: 0.90–2.17) for arrhythmia end points, and 1.15 (95% CI: 0.78–1.71) for these end points combined (Figure 1). Taken together, PKP2 c.419C>T genotype did therefore not predict heart failure or arrhythmia end points, individually or combined (Figure 1; P-values: 0.14–0.48).

Risk of heart failure, arrhythmia, combined end point (heart failure or arrhythmia), and all-cause mortality by PKP2 c.419C>T genotype in the general population. Ratios were multifactorially adjusted for age, gender, total cholesterol, body mass index, alcohol consumption, smoking, hypertension, diabetes, and ischemic heart disease at baseline. Heart failure end points: included heart failure/cardiomyopathy (n=1102 non-carriers; n=14 carriers), dyspnea/edema (n=410 non-carriers; n=3 carriers), and myocarditis (n=5 non-carriers; n=0 carriers). Arrhythmia end points included tachyarrhythmia (n=1.273 non-carriers; n=16 carriers), syncope (n=480 non-carriers; n=7 carriers), cardiac arrest (n=126 non-carriers; n=0 carriers).
The multifactorially adjusted HR for all-cause mortality was 1.33 (95% CI: 0.98–1.80) (Figure 1; P=0.07). Longevity, that is, the proportion surviving as a function of age was similar in carriers and non-carriers (Figure 2; log-rank P=0.14), and the median survival time was, respectively, 77.8 and 80.7 years in carriers and non-carriers (P=0.09).

Cumulative survival as a function of age and Plakophilin-2 c.419C>T genotype in the general population.
Case–control study
Characteristics of the 517 unrelated patients referred for genetic testing and their age- and sex-matched controls are shown in Table 3. The case group had a slightly lower median age (51 versus 54 years) caused by the case group including 23 children (age <18 years); however, none of the children were c.419C>T carriers and exclusion of this subgroup did not change the overall conclusions. Frequencies of c.419C>T were as follows: ARVC, 3/114 or 2.6% (0.5–7.5%); HCM/DCM, 2/316 or 0.6% (0.08–2.3%); LQTS, 0/87 or 0.0% (0.0–4.2%); or combined, 5/517 or 0.97% (0.3–2.2%). No patients with long QT syndrome were c.419C>T carriers. In carriers compared with non-carriers, odds ratios were 2.11 (0.50–8.99; P=0.31) for ARVC, 0.72 (0.16–3.28; P=0.68) for HCM/DCM, and 1.28 (0.46–3.54; P=0.64) for all cases combined (Figure 3). Furthermore, one proband with ARVC, and both probands with HCM carried other disease-associated/causing variants in, respectively, DSC2 c.4G>A (p.(E2K)), MYBPC3 c.2774_2775delAG (p.(E925Afs1049X)) and MYBPC3 c.906-36 A>G (premature stop codon in exon 12). Finally, c.419C>T did not segregate with disease in two families where cosegregation analysis was possible (data not shown).

Risk of arrhythmogenic right ventricular cardiomyopathy (ARVC), hypertrophic (HCM) and dilated cardiomyopathy (DCM), and the combined end point by PKP2 c.419C>T genotype in 517 unrelated individuals referred for genetic testing. No carriers were identified among 87 patients with long QT syndrome.
Taken together, data on intermediate parameters and prospective risk in the CCHS, as well as data from the case–control study, do not support an effect of c.419C>T on risk of heart failure or arrhythmia end points, or on risk of ARVC, HCM/DCM, or LQTS.
In vitro cell studies
Immunofluorescence experiments showed that wild-type and mutant fusion protein GFP-PKP2 were both correctly localized at the intercalated discs (Figure 4a). In some cells, the overexpressed GFP-PKP2 was also localized in intracellular vesicles, but their amount and localization were similar in wild-type and mutant transfected cells. In recombinant PKP2-expressing transfected cells, investigation of the junctional proteins plakoglobin, N-cadherin and connexin-43 through specific immunolabelling showed similar findings in all cells tested, indicating preserved cellular junction.

In vitro characterization of the PKP2 c.419C>T variant. (a) Rat neonatal cardiomyocytes transfected with either wild-type (WT) or PKP2 c.419C>T pEGFPN3-PKP2. PKP2GFP-fusion proteins (green labeling) are localized at the intercalated disks and sometimes in intracytoplasmic vesicles. The other junctional proteins plakoglobin, connexin-43, and N-cadherin are normally localized at the intercalated discs (red labeling). All x40 magnification. (b) In proliferation assays, HEK293T cells were transfected with either WT, PKP2 p.S140F pEGFPN3-PKP2 (p.S140F), or empty pEGFPN3 (GFP). BrdU−/cells+ (control well with non-transfected HEK293T− cells without the addition of BrdU) and BrdU+/cells− (control well with the addition of BrdU without HEK293T cells) bars represent background noise. Bromodeoxyuridine (BrdU) incorporation was measured with ELISA. Results are shown as±s.e.m. NS, non-significant (P=0.63).
In proliferation experiments, the level of BrdU incorporation was measured in HEK293T cells transfected with wild-type-PKP2-GFP, mutant PKP2-GFP, or GFP alone (Figure 4b). Levels of incorporation of BrdU did not differ between cells transfected with wild-type or mutant PKP2-GFPs (P=0.63).
Evolutionary conservation and predicted effect of PKP2 p.S140F
Multisequence alignment of orthologous PKP2 sequences is shown in Supplementary Figure 1, together with the predicted effect on protein function of c.419C>T. The variant was not conserved in mammals, for example, cow (Bos taurus) has an F in position 140. Furthermore, p.S140F was predicted to be, respectively, benign, tolerated, and a polymorphism by in silico prediction programs.
Discussion
The main findings of this study are that PKP2 c.419C>T, a variant which we identified in 0.94% of the general population (98/10 407 individuals), did not predict risk of heart failure, arrhythmia, mortality, or overall survival, and did not associate with electrocardiographic and echocardiographic changes, or levels of pro-BNP. Furthermore, c.419C>T genotype also did not associate with risk of ARVC, HCM/DCM, or long QT syndrome in our case–control study, and also did not segregate with disease in available families. Finally, in vitro data showed an indistinguishable immunosignal between cultured rat cardiomyocytes transfected with wild-type or mutant protein for four intercalated disc proteins. Taken together, these data suggest that PKP2 c.419C>T is neither a disease-causing nor a functional variant.
The clinical hallmark of ARVC is ventricular tachyarrhythmias causing palpitations, syncope, and cardiac arrest. The clinical course of ARVC is variable, but often characterized by arrhythmic hot phases,21 which may mimic myocarditis. Typical symptoms of DCM include dyspnea and edema. We encompassed these disease manifestations in diagnoses drawn from national registries and compared the incidences of myocarditis, heart failure/cardiomyopathy, tachyarrhythmia, cardiac arrest, syncope, and dyspnea/edema in PKP2 c.419C>T carriers and non-carriers without any significant association. In addition, as both ARVC and DCM patients often exhibit ECG abnormalities,22, 23 including arrhythmias, ventricular conduction abnormalities, and abnormal electrocardiographic intervals, we investigated the association of PKP2 c.419C>T genotype with these parameters, and again found no associations. These data were consistent with a lack of echocardiographic signs of left ventricular systolic dysfunction or chamber dilatation – findings that contribute to the definition of DCM. Derivates of BNP are well-established, sensitive markers of heart failure, and myocardial stress, but no difference was found between carriers and non-carriers.
The PKP2 p.S140F variant is located in the N-terminal part of plakophilin-2, which has been reported to be of particular importance for proper cellular localization.24 Our cellular experiments did not document any difference in the expression of plakophilin-2, plakoglobin, N-cadherin, or connexin-43 in cells transfected with wild-type or mutant PKP2. Reduced levels of plakoglobin and connexin-43 have been reported as a very common finding in patients with ARVC, regardless of the underlying genetic etiology.25, 26 Our findings suggest that PKP2 c.419C>T does not alter the localization of the four investigated proteins. Plakophilin-2 has also been implicated in cellular proliferation of epicardial cells27 and an ARVC-associated variant in plakoglobin has been shown to increase the proliferation rate in HEK293 cells.28 We therefore performed proliferation assays but did not find any effect of the PKP2 c.419C>T variant on BrdU incorporation suggestive of a lack of functional effect of PKP2 c.419C>T.
Our finding that ~1% of the population carry the PKP2 c.419C>T variant is much higher compared with expected by disease prevalence, assuming a disease-causing effect of the variant, a monogenic mode of inheritance, and a penetrance between 25 and 100%. DCM and ARVC have estimated prevalences of ~1:2500 and ~1:5000,29 respectively, and a very heterogeneous genetic background with more than 50 different genes associated with the two diseases. However, little is known about the prevalence of cardiomyopathy-associated variants in the general population. A single study30 has investigated the role of five ARVC-associated desmosomal variants (four missense and one deletion) in a Finish general population sample. The combined prevalence of these variants was 0.5%. However, the Finish population is a known genetic isolate and very limited phenotypic data were available, hampering the establishment of clear disease association. Sequencing of five ARVC-associated genes in 427 apparently healthy controls identified rare missense variants in 16% of the controls, suggesting a high frequency of benign genetic variation in the desmosomal genes.31 Data from the Washington Exome Server show a prevalence of the PKP2 c.419C>T variant of 25 out of 8575 alleles (0.3%) of European American ancestry and only of 1 out of 4405 alleles (0.02%) of African American ancestry. Combined with our data, this suggests that PKP2 c.419C>T displays geographical and ethnical variation.
Our study has the following limitations: no specific echocardiographic evaluation of the right ventricle, no MRI data available, and lack of data regarding specific ARVC-associated electrocardiographic findings (e.g. epsilon or negative T-waves). Furthermore, no data on the family history or myocardial histology of the PKP2 c.419C>T carriers were available. Although our population study is very large and has a high event rate, it is not possible to completely rule out a very small effect of the variant that would have been unmasked if the study had been even larger. Our in vitro experiments also have limitations: a single genetic variant is introduced into a foreign cell with a different genetic background, which may not be representative of the in vivo environment, and no testing of protein interactions.
In conclusion, we identified PKP2 c.419C>T in ~1% of the general population. Combining epidemiological data, with data on patients referred for genetic testing for ARVC or HCM/DCM, and data from in vitro studies, PKP2 c.419C>T did not associate with heart failure, arrhythmias, or premature death, with ARVC or HCM/DCM, or with effects in vitro, suggesting that this is not a disease-causing variant.
References
Sen-Chowdhry S, McKenna WJ : Sudden death from genetic and acquired cardiomyopathies. Circulation 2012; 125: 1563–1576.
Bhuiyan ZA, Jongbloed JD, van der SJ et al: Desmoglein-2 and desmocollin-2 mutations in Dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet 2009; 2: 418–427.
Den Haan AD, Tan BY, Zikusoka MN et al: Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet 2009; 2: 428–435.
Garcia-Pavia P, Syrris P, Salas C et al: Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 2011; 97: 1744–1752.
Elliott P, O’Mahony C, Syrris P et al: Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 314–322.
Christensen AH, Benn M, Bundgaard H, Tybjaerg-Hansen A, Haunso S, Svendsen JH : Wide spectrum of desmosomal mutations in Danish arrhythmogenic right ventricular cardiomyopathy patients. J Med Genet 2010; 47: 736–744.
Xu T, Yang Z, Vatta M et al: Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2010; 55: 587–597.
Christensen AH, Benn M, Tybjaerg-Hansen A, Haunso S, Svendsen JH : Missense variants in plakophilin-2 in arrhythmogenic right ventricular cardiomyopathy patients – disease-causing or innocent bystanders? Cardiology 2010; 115: 148–154.
Syrris P, Ward D, Asimaki A et al: Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 2006; 113: 356–364.
Gerull B, Heuser A, Wichter T et al: Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004; 36: 1162–1164.
Dalal D, Molin LH, Piccini J et al: Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation 2006; 113: 1641–1649.
Fressart V, Duthoit G, Donal E et al: Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 2010; 12: 861–868.
Sen-Chowdhry S, Syrris P, Prasad SK et al: Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008; 52: 2175–2187.
Groeneweg JA, van der Zwaag PA, Jongbloed JDH et al: Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm 2013; 10: 548–559.
Marcus FI, McKenna WJ, Sherrill D et al: Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31: 806–814.
Johannsen TH, Frikke-Schmidt R, Schou J, Nordestgaard BG, Tybjærg-Hansen A : Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J Am Coll Cardiol 2012; 60: 2041–2048.
Prineas R, Crow R, Blackburn H : The Minnesota Code Manual of Electrocardiographic Findings. Littleton, MA, USA: John Wright-PSG Inc, 1982.
Lang RM, Bierig M, Devereux RB et al: Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005; 18: 1440–1463.
Goetze JP, Mogelvang R, Maage L et al: Plasma pro-B-type natriuretic peptide in the general population: screening for left ventricular hypertrophy and systolic dysfunction. Eur Heart J 2006; 27: 3004–3010.
Duboscq-Bidot L, Xu P, Charron P et al: Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res 2008; 77: 118–125.
Sen-Chowdhry S, Syrris P, McKenna WJ : Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2007; 50: 1813–1821.
Peters S, Trummel M : Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: value of standard ECG revisited. Ann Noninvasive Electrocardiol 2003; 8: 238–245.
Xiao HB, Roy C, Fujimoto S, Gibson DG : Natural history of abnormal conduction and its relation to prognosis in patients with dilated cardiomyopathy. Int J Cardiol 1996; 53: 163–170.
Chen X, Bonne S, Hatzfeld M, van RF, Green KJ : Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem 2002; 277: 10512–10522.
Fidler LM, Wilson GJ, Liu F et al: Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med 2009; 13: 4219–4228.
Asimaki A, Tandri H, Huang H et al: A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009; 360: 1075–1084.
Matthes SA, Taffet S, Delmar M : Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun Adhes 2011; 18: 73–84.
Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ : A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2007; 81: 964–973.
Raju H, Alberg C, Sagoo GS, Burton H, Behr ER : Inherited cardiomyopathies. BMJ 2011; 343: d6966.
Lahtinen AM, Lehtonen E, Marjamaa A et al: Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2011; 8: 1214–1221.
Kapplinger JD, Landstrom AP, Salisbury BA et al: Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol 2011; 57: 2317–2327.
Acknowledgements
We thank the participants of the Copenhagen City Heart Study for their willingness to participate.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Christensen, A., Kamstrup, P., Gandjbakhch, E. et al. Plakophilin-2 c.419C>T and risk of heart failure and arrhythmias in the general population. Eur J Hum Genet 24, 732–738 (2016). https://doi.org/10.1038/ejhg.2015.171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.171
This article is cited by
-
Arrhythmogenic Cardiomyopathy: from Preclinical Models to Genotype–phenotype Correlation and Pathophysiology
Stem Cell Reviews and Reports (2023)
-
A Systematic Analysis of the Clinical Outcome Associated with Multiple Reclassified Desmosomal Gene Variants in Arrhythmogenic Right Ventricular Cardiomyopathy Patients
Journal of Cardiovascular Translational Research (2023)
