
Abstract
Isolated populations are valuable resources for mapping disease genes, as inbreeding increases genome-wide homozygosity and enhances the ability to map disease alleles on a genetically uniform background within a relatively homogenous environment. The populations of Daghestan are thought to have resided in the Caucasus Mountains for hundreds of generations and are characterized by a high prevalence of certain complex diseases. To explore the extent to which their unique population history led to increased levels of inbreeding, we genotyped >550 000 autosomal single-nucleotide polymorphisms (SNPs) in a set of 14 population isolates speaking Nakh-Daghestanian (ND) languages. The ND-speaking populations showed greatly elevated coefficients of inbreeding, very high numbers and long lengths of Runs of Homozygosity, and elevated linkage disequilibrium compared with surrounding groups from the Caucasus, the Near East, Europe, Central and South Asia. These results are consistent with the hypothesis that most ND-speaking groups descend from a common ancestral population that fragmented into a series of genetic isolates in the Daghestanian highlands. They have subsequently maintained a long-term small effective population size as a result of constant inbreeding and very low levels of gene flow. Given these findings, Daghestanian population isolates are likely to be useful for mapping genes associated with complex diseases.
Similar content being viewed by others

Introduction
Human consanguinity has been shown to lead to increased rates of recessive genetic disorders and has likely had a significant negative effect on polygenic traits influencing health.1, 2, 3, 4 An effect of consanguineous mating recognizable in most organisms is inbreeding depression, defined as the reduced survival and fertility of offspring of related individuals.5 A plausible explanation for inbreeding depression is increased homozygosity for partially recessive deleterious mutations (partial dominance). This explanation is currently favored over the alternative of increased homozygosity for alleles at loci with heterozygote advantage (overdominance).6 Within humans increased homozygosity is most likely to be observed in isolated populations. In fact the identification of rare recessive disease genes is greatly facilitated by conducting mapping studies in isolated populations founded by a limited number of individuals in whom causative rare alleles are often observed in homozygous form. In addition, population isolates are hypothesized to possess lower levels of allelic heterogeneity underlying disease traits and higher levels of linkage disequilibrium (LD), which suggests, when coupled with a relatively homogeneous background, that they may also be useful for the identification of susceptibility genes for complex diseases and quantitative traits.7, 8, 9, 10
However, it is important to characterize the level of inbreeding in a population isolate to determine its usefulness for examining particular diseases. Whereas the study of population isolates that were recently founded from a small number of individuals and that have undergone population expansions are useful for identifying loci associated with rare recessive diseases (for example, Finns), older population isolates with constant small Ne may be more helpful for finding loci contributing to complex disease (for example, the Saami).11 Ethnographic pedigree estimates of inbreeding are usually limited in resolution to three to six generations,3 whereas unknown relationships are more likely to exist in populations with higher rates of consanguinity. Fortunately, the availability of dense genome-wide genetic data now makes it possible to characterize the level of population inbreeding over much larger timescales.
The traditional coefficient of inbreeding, F, is estimated on the basis of the observed versus expected number of homozygous genotypes in a population, with the latter derived from population allele frequencies. However, F statistics will undervalue the true level of inbreeding if the level of homozygosity is high.12 An alternative method for inferring levels of inbreeding that is less sensitive to biases resulting from estimating population allele frequencies is to examine the length distribution of Runs of Homozygosity (ROH) within individuals.13 Importantly, this method can also distinguish between recent and more ancient consanguinity based on homozygous tract lengths.10, 12, 14, 15, 16, 17, 18, 19, 20 Recently, a model-based approach for detecting ROH was described that better accounts for missing data because of potential sequencing error, is potentially more sensitive to detecting true autozygous segments and does not require a priori size boundaries for different size classes that may introduce bias when comparing populations with different histories.19, 21, 22
In this study we examine the level of inbreeding and population structure using genome-wide autosomal single-nucleotide polymorphisms (SNPs) genotyped in 20 Daghestanian populations belonging to 19 ethnic groups from the Caucasus Mountains. Of the 50 indigenous Caucasian groups inhabiting the region today, 26 live within Daghestan, a territory comprising 50 300 km2 of the North Caucasus. Archaeological sites first appear in the Mesolithic (~10 000 BP) after the retreat of glaciers, with evidence of stable continuous human occupation thereafter.23, 24 No archaeological or linguistic evidence points to further major movements into the highlands.24, 25, 26, 27, 28 Thus, populations may have lived in the same highland region for hundreds of generations in relative isolation.24, 29 Consistent with this hypothesis, modern highland Daghestanian groups speak Nakh-Daghestanian (ND) or Northeast Caucasian languages that fall into deeply divergent branches of a single language family that appears to be endemic to the eastern Caucasus.25, 26, 27 Moreover, some Daghestanian isolates are characterized by a high prevalence of a number of genetic diseases, including schizophrenia, major recurrent depression, neuromuscular dystrophy, cardiovascular diseases and autosomal recessive deafness.30, 31, 32, 33, 34, 35, 36 Limited pedigree data suggest very high mean inbreeding coefficients (0.005–0.0134),32, 37 whereas population genetic studies have been restricted to autosomal Alu insertion and STR markers, the Y chromosome and mtDNA.32, 37, 38, 39, 40, 41, 42, 43, 44, 45
Materials and methods
Sample collection
Cheek swabs were obtained from 613 individuals from 19 ethnic groups living in 20 villages. Figure 1 shows a map of the sampled region, and Supplementary Table 1 lists samples defined by language and sample sites. Note that 14 of the Daghestanian populations speak unique languages that are part of the ND language family, whereas 6 speak non-ND languages. Sampled individuals were thought to be unrelated for at least three generations. Informed consent was obtained from all individuals according to protocols approved by the University of Arizona Human Subjects Committee. Armenian samples were collected by LY in Ararat Region, Armenia with a written consent form approved by the Institute of Molecular Biology, Yerevan, Armenia.

Approximate geographic location of sampling sites. Of the 20 Daghestanian populations sampled here, 14 speak unique languages that are part of the ND language family. Of the remaining six populations (non-ND), three speak languages that are closely related to the Turkic branch of Altaic language family (Kumyks, Nogais and Azerbaijans), and three speak languages belonging to the Iranian language branch of the Indo-European language family (ethnic Tats, Mountain Jews and a group of Azerbaijans originated in Iran). See Supplementary Table 1 for population codes. ND and non-ND populations are shown in circle and triangle, respectively.
Genotyping and curation of autosomal SNPs
We genotyped 567 096 SNPs in 314 samples from Daghestan using the Affymetrix (Axiom, Santa Clara, CA, USA) platform, as well as in an additional 261 samples from the Caucasus (that is, non-Daghestanian), Near East, Europe, Central Asia and South Asia (Supplementary Table 1). Data were submitted to the NCBI/dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=1061909. Submitter Batch ID: Daghestan_SNP_Affymetrix. Our Axiom-genotyping data can be also accessed through our website at http://hammerlab.biosci.arizona.edu/SupplementaryData/HAMMER_LAB_AFFYMETRIX_CHIP.tar. Other than the Armenian samples, which are described here for the first time, all of the additional non-Daghestanian samples were included in previous studies.46, 47 Only SNPs located on the 22 autosomes (with centromeric regions removed) spanning a total length 2738 Mb were included in downstream analyses. We used the software PLINK1.0713 to filter the data set by removing SNPs with 10% or more missing genotypes and SNPs with a minor allele frequency (MAF) less than 1%. We also removed SNPs in high LD (window size: 50 SNPs; sliding window: 5 SNPs; r2 threshold=0.8), leaving a panel of 549 008 SNPs. Only a single individual from any pair of samples demonstrating high relatedness within the same population (inferred as third-degree relatives or higher using a PLINK PI_HAT value of ≥0.125) was included in the analysis. After removing close relatives, the total number of samples in our Axiom data set was 480. For several analyses (principal component analysis (PCA), ADMIXTURE, AMOVA, estimation of traditional coefficient of inbreeding, F), we intersected our data with publicly available samples (Supplementary Table 1). After applying the same filtering described above and removing obvious PCA outliers, the final merged data set resulted in 104 519 SNPs for 1100 individuals across 56 populations.
Data analysis
To evaluate the putative genetic ancestry of Daghestanian ethnic groups, we conducted a PCA using SMARTPCA48 and ADMIXTURE analysis49 on the merged autosomal data set of our samples from Daghestan, the Caucasus, Near East, Europe, Central Asia and South Asia. To minimize bias from high within-group covariance for larger samples sizes, we used a ‘drop one in’ procedure described by Veeramah et al.10 Genetic differentiation was estimated by the ARLEQUIN 3.5 software50 and SMARTPCA program in the EIGENSOFT software package.48
To assess the extent of inbreeding in Daghestan, we used three different measures of genome-wide homozygosity: (1) the coefficient of inbreeding F as obtained from PLINK,13 (2) a population-based estimate of the distribution of ROH as measured by a logarithm of the odds (LOD) score19, 22 with the same parameters as those used by Pemberton et al19 and (3) an individual-based estimate of the distribution of ROH using PLINK13 with the parameters identical to McQuillan et al.20 Estimates of F as obtained from PLINK (which assumes independence among SNPs) were performed on the merged data set of 104 519 SNPs in 56 populations, whereas the LOD and PLINK-ROH analyses were performed on our Axiom data set in 480 individuals from 33 populations (549 008 SNPs). To explore whether Daghestanian populations differ in patterns of ROH from surrounding populations, for PLINK-ROH analysis we supplemented our data set with seven Caucasus groups (N=132) from published data genotyped on different Illumina platforms: Adygei, Georgians, Lezgins, Abkhasian, Balkar, Chechen and North Ossetian (Supplementary Table 1). The number and density of SNPs included in ROH analyses varied only marginally across data sets: 549 008 and 4.9 kb/SNP (this study), 509 119 and 5.3 kb/SNP,45 510 154 and 5.3 kb/SNP51 and 569 388 and 4.7 kb/SNP.52, 53 To investigate the effect of potential ascertainment bias on the genome-wide homozygosity, we repeated principle PLINK-ROH analyses removing SNPs with a MAF less than 10%, leaving a panel of 380 015 SNPs. Applying different MAF thresholds of 10% changed the absolute values of ROH statistics, but had little effect on the relative order of populations and/or individuals (Supplementary Figure 1). The ROH frequencies and ROH similarities between populations were calculated as suggested in Pemberton et al.19 The Pearson correlation coefficient among total lengths in different pairs of classes was estimated using the R package.54
We evaluated the decay of LD with recombination distance for each chromosome using the genotypic-based r2 statistic estimated in PLINK. ND-speaking populations were compared with our European and Yoruba (YRI) reference populations from the HapMap collection. The data set was filtered to remove SNPs with 10% or more missing genotypes and SNPs with MAF less than 5%, leaving a panel of 538 788 SNPs. To control for the effect of sample size differences on r2, we randomly re-sampled 11 individuals when computing r2 for each SNP pair.55 All SNPs were assigned to genetic map positions using the CEU or YRI HapMap (hg18) recombination maps. r2 values were binned into 50 genetic distance groups between 0.005 and 0.25 cM in increments of 0.005 cM, and their mean r2 was used for further calculation of effective population size (Ne). For each population with sample size N≥10 we estimated Ne=1/(4c) × ((1/r 2)−2), where r2 was adjusted for sample size: (r2–1/n) and c is the recombination distance between loci in Morgans56, 57 (1 cM≈1.1-1.2 Mb). The standard error and confidence intervals for Ne were derived from separate analysis of each autosome.56 As recombination distance between markers is approximately inversely proportional to the number of generations t≈1/(2c),58 we estimated Ne in different time intervals assuming 25 years per generation.
Results
Genetic structure of Daghestanian populations
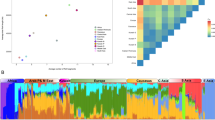
PCA revealed relatively distinct clusters of Europeans, South Asians and Central Asians, whereas Daghestanian samples (except the Nogais and Mountain Jews) are intermingled with other individuals from the Caucasus and show an affinity with our Near Eastern samples (Figure 2). Among our Near Eastern groups, Turks and Iranians demonstrate genetic similarity to populations from Daghestan, in particular, to Azerbajans from Daghestan (who consist of Turkic and Iranian speakers) and Tats (who are recent migrants from Persia). In general, the clustering within and general relatedness between populations corresponds approximately to their relative geographical location. We applied a STRUCTURE-like approach49 to estimate individual ancestry in K hypothetical ancestral populations. The best projecting accuracy was observed for a model with K=7 (Supplementary Figure 2). Overall, this approach revealed population structure consistent with the PCA analysis. For K1–K5 the ancestry proportions for all Daghestanian populations (except the Nogais and Mountain Jews) were similar to other populations from the Caucasus, Iranians and the Turks (Supplementary Figure 3). With higher K values, a single ancestry component emerged in the Hinukh, Hunzib and Tsez populations, whereas a separate ancestry component prevailed in the Akhvakh, Ratlub and Tindal populations.

PCA analysis using the ‘drop one in’ technique for 56 populations. Circled positions indicate the median coordinate values for populations. See Supplementary Table 1 for population codes.
The mean pairwise FST value for our 19 Daghestanian ethnic groups was 0.0174 (Table 1). When only the 13 ND-speaking populations were included in the analyses, the FST estimate increased to 0.0186. Despite the small geographic area involved, this value is significantly higher than that observed among our Caucasian, Near Eastern, European, Central Asian or South Asian samples, although a strict comparison is difficult as the majority of the latter samples are cosmopolitan (that is, may be from multiple subpopulations).
Inbreeding coefficient and ROH in Daghestanian populations
Population samples from the Caucasus, Europe and Central Asia demonstrate the lowest F values (mean 0.0017, 0.0029 and 0.0113, respectively; Supplementary Table 2 and Supplementary Figure 4). Our Near Eastern and South Asian samples are characterized by F values that are two to three times higher. Non-ND ethnic groups from Daghestan show low F values similar to other populations from the Caucasus. On the other hand, ND-speaking populations reveal significantly higher coefficients of inbreeding compared with populations from other geographical regions (mean 0.0447, P<0.0001).
To examine ROH at the population level, we applied the Guassian mixture likelihood approach of Pemberton et al,19 which incorporates population allele frequencies to estimate a LOD score for measuring how likely it is for a segment to be autozygous.19, 21, 22 All ROH were subdivided into three classes: short (A), intermediate (B) and long (C) with a model-based clustering algorithm (with longer runs generally attributed to more recent inbreeding). LOD score thresholds and the size boundaries between classes A and B and classes B and C for each population are presented in Supplementary Table 3. The bimodal distribution of LOD scores (Supplementary Figure 5) demonstrates an increased presence of autozygous genomic regions in our samples from Daghestan and the Near East compared with those from the Caucasus, Europe, Central Asia and South Asia. The population-specific thresholds show negative values for five ND populations and Saudi Arabians (Supplementary Table 3), suggesting that autozygosity is more common in these populations. The ND populations also demonstrated significantly higher boundary sizes for classes A and B, and B and C than those from any other geographical regions suggesting longer ROH sizes on average in all three classes. The difference is most notable in the total length of ROH in class C and in all three classes combined (Figure 3). The variance appears to be higher in Daghestan, which may be suggestive of a lower Ne.12

Distribution of total length of different ROH classes per individual in 40 populations. A box plot and a kernel density plot are shown as ‘violin plots’ for classes A (a), B (b), C (c) and A, B, C classes pooled together (d). The white horizontal line is the median, whereas the black bar represents the interquartile range. Populations are grouped according to geographic regions.
In an effort to distinguish whether the observed distribution of different size classes of ROH in various geographic regions may have arisen via distinctive demographic and/or evolutionary processes, we calculated the correlations of distance matrices based on the frequencies of different ROH classes and distance matrices based on frequencies of autosomal SNPs (N=549 008). Autosomal SNP FST values show a strong and highly significant correlation with distances based on the frequencies of ROH in classes A (r=0.601, P=0.007) and B (r=0.575, P=0.004), but no correlation with class C ROH (r=−0.066, P=0.705). We then repeated pairwise FST analysis excluding SNPs from different ROH classes. After removing SNPs from ROH classes C, B and C, and A and B and C, the average differentiation for ND-speaking populations decreases from 0.0254 to the values of 0.0223, 0.0203 and 0.0182, respectively. This is by far the largest absolute reduction for any regional group for either category. This suggests that much of the excessive population differentiation observed above is related to more prominent long-term inbreeding in Daghestanian populations (Supplementary Table 4).
Finally, we inferred levels of inbreeding at the level of individuals by examining the length distribution of ROH in PLINK (that is, unlike for F and the model-based ROH approach, this analysis is not reliant on population allele frequency estimation). In the expanded sample of 612 individuals, the total ROH length and ROH number per individual ranges from 5.5 to 465.7 Mb and from 7 to 186, respectively. The population mean of the cumulative ROH length per individual varies from 16.29 Mb (SE=2.96 Mb) in our sample of Sri Lankans to 219.3 Mb (SE=37.0 Mb) in the Hinukhs from Daghestan. Similarly, the population mean of the ROH number per individual shows the minimum value of 19.1 (SE=2.82) in the Sri Lankans, with the maximum reaching 108.8 (SE=25.53) among Hinukhs (Supplementary Table 5, Supplementary Figure 6). The proportion of different length ROH fluctuates considerably among geographic regions. Samples from Daghestan, particularly ND speakers, differ strikingly from those in the other geographical regions. The mean of the total ROH length per individual in our ND-speaking sample (128.1 Mb, SE=6.54) is significantly higher than in those from any other geographical region (P<0.0005). Approximately 55% of ND-speaking individuals possess a single contiguous ROH tract with length >100 Mb, and the average fraction of the genome in ROH is 4.7%. Out of the 13 ND-speaking populations analyzed, 6 populations (Akhvakh, Hinukh, Hunzib, Ratlub, Tindal and Tsez) exhibit a cumulative ROH length >100 Mb in more than 70% of individuals. Similarly, the number of ROH per individual is significantly higher among our ND-speaking samples compared with other geographical regions (73.41, SE=2.09; P<0.0001).
Interestingly, substantially elevated ROH numbers and lengths are not seen in our non-ND-speaking population samples, with both parameters being similar to other populations from the Caucasus. As shown previously17, 59, 60 geographical regions differ in their frequency of distinctive pre-defined ROH length categories (Figure 4,Supplementary Figure 6). Whereas shorter ROHs (<2 Mb) are common in all regions, the longer ROHs are increasingly prevalent only in our ND-speaking samples and those from the Near East.

Regional distribution of ROH. The mean total ROH length (Mb)±SE is plotted for each geographic region.
We compared several ROH parameters in ND-speaking populations with those from other studies of genetic isolates.14, 15, 20, 61 The distribution of ROHs according to their size (in Mb; Supplementary Figure 7, Supplementary Table 6) clearly shows a higher proportion of individuals with extended regions of autozygosity in ND-speaking populations compared with individuals in other genetic isolates. For example, with 1.5 Mb used as the minimum length, endogamous Dalmatians and Orcadians had the mean ROH length of 35 and 28 Mb, respectively,20 and six geographically isolated villages in northeastern Italy were characterized by an average ROH of ~47 Mb.15 All of our ND-speaking populations are distinguished by a much higher mean ROH length of 92.6 Mb (that is, ranging from 50.4 Mb in Andians to 162.3 Mb in Hunzibs; Supplementary Table 6). When ROHs greater than 1 Mb are considered, the median ROH number and total ROH length for Daghestanian ND populations are 36.8 and 97.7 Mb, respectively, compared with ~16–19 and ~40–60 Mb for three isolates from Finland (Supplementary Table 6).61 One likely cause of this higher level of autozygosity is increased recent inbreeding among ND-speaking Daghestanian populations.
We evaluated hotspots in ND populations and contrasted them with those previously identified in European populations. Considering genomic regions of ≥50 adjacent SNPs, a total of 150 regions were identified as ROH hotspots (with minimum ROH frequencies higher than 10%) in ND populations (Supplementary Table 7). Among these 150 hotspots, 18 have been recognized in previous surveys of ROH in European populations18 and in worldwide human populations.19 Interestingly, 11 hotspots overlapped with 108 recently published schizophrenia-associated genetic loci.62
Estimation of effective population sizes
To further assess their level of genetic isolation, we evaluated the decay of LD over recombination distance for ND-speaking populations and compared this with our reference populations from Europe and YRI. We explored spatial and temporal variation in Ne estimates, although an exact comparison is not straightforward because the majority of the European samples may come from multiple subpopulations. All ND-speaking populations except the Avars show a significantly elevated level of LD at all recombination distances (Supplementary Figure 8). The Avars, the most numerous ND-speaking population, appear to have a comparable level of LD as European populations. Ne estimates vary significantly across populations in our study (Figure 5,Supplementary Table 8). The Ne value for YRI (12 113, SD=867) is slightly lower than that estimated in McEvoy et al.56 (13 900). The highest average estimate of Ne by region is found in our South Asian samples (Ne=10 365; SD=453). The highest Ne values for Daghestanian populations are observed in the Kumyks and Avars, which represent the two largest populations in Daghestan by census size. The smallest Ne values are found in ND-speaking populations (ranging from ~2730 to 4150) with the highest level of inbreeding (Akhvakh, Hunzib, Ratlub, Tindal and Tsez). (Note, that the Hinukh sample was omitted from this analysis because of small sample size). The average Ne for ND-speaking populations (4675, SD=1463) is approximately a half of the estimated Ne of our Near Eastern and European samples. The Ne estimates are significantly correlated with census population sizes in Daghestan (r=0.55, P=0.011), particularly for ND-speaking populations (r=0.72, P=0.009). LD observations in the bins between 0.005 and 0.25 cM in all populations allowed us to examine temporal variation in Ne from ~125 000-5000 years ago. The higher levels of LD over recombination distances in ND-speaking populations made it possible to extend our calculations between 0.005 and 0.36 cM, providing estimates of Ne to 3000 years ago. We chose European, ND-speaking and YRI populations to trace temporal variation of Ne (Supplementary Figure 9). Consistent with the earlier study56 we found a relatively large Ne for YRI, with a slight increase around 9000 years ago, and a population size expansion for Europeans for the last 10 000–12 000 years. All ND-speaking populations except the Avars show a steady decline in their effective population sizes.

The harmonic mean of Ne for each population with sample size N≥10 over 50 recombination distance classes between 0.005 and 0.25 cM with increment 0.005 cM. The SEs for Ne indicated by the bars were derived from separate analysis of each autosomal chromosome. Yorubans were included for comparisons with published data.
Discussion
Our study represents the first attempt to characterize genetic diversity in Daghestanian populations using high-density SNP analyses in a large number of ethnic groups (N=19). Marriage customs favor endogamy on both sides in all ND-speaking groups, which vary in total population size from ~600 individuals in Hinukhs to 785 300 in Avars. Traditional consanguineous marriages are commonly between cousins (that is, mostly between paternal cousins) in order to keep resources within the clan (Tukhum). As a result, ethnic identity is preserved, and numerous long-isolated, inbred small populations are created.39, 63, 64, 65 Limited pedigree data suggest very high inbreeding coefficients (0.005–0.0134). Three different measures of genome-wide homozygosity consistently reveal the highest level of inbreeding in Daghestanian ND-speaking groups compared with our Eurasian population samples. Our ND-speaking sample has the highest traditional coefficient of inbreeding F, the longest genomic stretches of homozygosity for all ROH length categories and a higher proportion of individuals with extended regions of autozygosity.
The extent of recent inbreeding in Daghestanian populations is evidenced by the presence of very long ROH segments, with a large proportion of individuals in ND-speaking population possessing segments >100 MB in length (Figure 4, Supplementary Figure 7,Supplementary Table 5). To put this number in a temporal context, under a very simplistic model, a mean ROH length of 10 and 25 cM, respectively, would be compatible with the parents sharing a common ancestor five and two generations ago.66 However, we note that the scenario for Daghestan is likely much more complicated, with multiple layers of possible inbreeding. Although a high coefficient of inbreeding, F, and the presence of long autozygous segments are consistent with previous inferences of inbreeding coefficients estimated from pedigree data of up to 11–14 generations (275–350 years),32, 37 our combined analysis demonstrates that inbreeding has likely been a common feature in Daghestan over a sustained period, with shorter runs indicative of more ancient inbreeding also more prominent in Daghestanian versus other populations.
In fact, our results suggest that Daghestan demonstrates the highest levels of autozygosity observed on mainland Eurasia to date.14, 15, 16, 18, 20, 60, 61, 67, 68 This effect appears to predominate particularly among the ND-speaking populations, with non-ND-speaking Daghestanian and non-Daghestanian Caucasus populations showing levels of autozygosity that are comparable to other Eurasian populations. In addition, a high level of genetic structure is observed among ND-speaking populations. The long-term Ne of these populations as estimated by the decay in LD appears to have been lower than other European populations for the last 10 000 years. These results are compatible with the hypothesis of long-term isolation of Daghestanian ethnic groups living in a highly mountainous region with extensive inbreeding and negligible gene flow among villages speaking different languages within the ND language family.
Our results are also compatible with the high incidence of various complex diseases in a number of Daghestanian populations.30, 32, 33, 34, 68 Several genetic studies of quantitative, disease-related phenotypes have already been successfully carried out in Daghestanian small isolated communities.30, 32, 33, 34, 69, 70, 71, 72, 73, 74 These studies observed notable within- and between-isolate diversity in clinical and genetic heterogeneity likely because of differences in founders, subsequent endogamy and inbreeding within the isolates. The results from these studies suggest that mapping genes of complex diseases, including major depressive disorder, schizophrenia, hearing loss across genetically homogeneous isolates, can facilitate detecting linkage signals and expedite the search for susceptibility genes when combined with the methods that identify structural genomic and nucleotide variation in linkage regions. This suggests that the populations of Daghestan are excellent candidates for studies of the effects of homozygosity on the health of subjects with a shared genetic and environment background, as well as for disease gene mapping. In particular, Daghestan appears to possess a relatively large number of what could be defined as ancient or primary population isolates.75 The utility of population isolates with low long-term Ne in the mapping and identification of genes is not only limited to the study of rare diseases—isolated populations also provide a useful resource for the identification of susceptibility genes for complex diseases, initial insights into disease pathogenesis and for understanding the biology underlying common diseases and their component traits.8, 76, 77, 78, 79, 80 The study of isolated populations facilitates the discovery of disease-associated genes with alleles that have increased in frequency as a result of a strong genetic drift.78 As such, the genetic isolates of Daghestan may provide the opportunity to identify both common and rare disease-causing variants through association studies.
References
Acevedo-Whitehouse K, Gulland F, Greig D, Amos W : Inbreeding: disease susceptibility in California sea lions. Nature 2003; 422: 35.
Bittles A : Consanguinity and its relevance to clinical genetics. Clin Genet 2001; 60: 89–98.
Bittles AH, Black ML : Evolution in health and medicine Sackler colloquium: consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA 2010; 107 (Suppl 1): 1779–1786.
Nalls MA, Guerreiro RJ, Simon-Sanchez J et al: Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer's disease. Neurogenetics 2009; 10: 183–190.
Charlesworth D, Willis JH : The genetics of inbreeding depression. Nat Rev Genet 2009; 10: 783–796.
Edmands S : Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol Ecol 2007; 16: 463–475.
Arcos-Burgos M, Muenke M : Genetics of population isolates. Clin Genet 2002; 61: 233–247.
Kristiansson K, Naukkarinen J, Peltonen L : Isolated populations and complex disease gene identification. Genome Biol 2008; 9: 109.
Sheffield VC, Stone EM, Carmi R : Use of isolated inbred human populations for identification of disease genes. Trends Genet 1998; 14: 391–396.
Veeramah KR, Tonjes A, Kovacs P et al: Genetic variation in the Sorbs of eastern Germany in the context of broader European genetic diversity. Eur J Hum Genet 2011; 19: 995–1001.
Terwilliger JD, Zollner S, Laan M, Paabo S : Mapping genes through the use of linkage disequilibrium generated by genetic drift: 'drift mapping' in small populations with no demographic expansion. Hum Hered 1998; 48: 138–154.
Keller MC, Visscher PM, Goddard ME : Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011; 189: 237–249.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Colonna V, Pistis G, Bomba L et al: Small effective population size and genetic homogeneity in the Val Borbera isolate. Eur J Hum Genet 2013; 21: 89–94.
Esko T, Mezzavilla M, Nelis M et al: Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur J Hum Genet 2013; 21: 659–665.
Gross A, Tonjes A, Kovacs P et al: Population-genetic comparison of the Sorbian isolate population in Germany with the German KORA population using genome-wide SNP arrays. BMC Genet 2011; 12: 67.
Leutenegger AL, Sahbatou M, Gazal S, Cann H, Genin E : Consanguinity around the world: what do the genomic data of the HGDP-CEPH diversity panel tell us? Eur J Hum Genet 2011; 19: 583–587.
Nothnagel M, Lu TT, Kayser M, Krawczak M : Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum Mol Genet 2010; 19: 2927–2935.
Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ : Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet 2012; 91: 275–292.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al: Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Broman KW, Weber JL : Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain. Am J Hum Genet 1999; 65: 1493–1500.
Wang S, Haynes C, Barany F, Ott J : Genome-wide autozygosity mapping in human populations. Genet Epidemiol 2009; 33: 172–180.
Kotovich VG, Sheikhov NB : Arkheologicheskoe izuchenie Dagestana za 40 let (Archeological Study of Dagestan during Last 40 Years): Uchenye Zapiski IIYaL Dagestan Filial AN SSSR (IIYaL Scientific Transections Dagestan Filial Acad. Nauk SSSR. Makhachkala: IIYaL, 1964; Vol 8.
Gadzhiev MG, Davudov OM, Shikhsaidov SM : Istoriya Dagestana (The History of Dagestan). Moscow: Nauka, 1996.
Nichols J : Language dispersal from the Black Sea region; in: Yanko-Holmbach V, Gilbert AS, Panin N, Dolukhanov PM (eds): The Black Sea Flood Question: Changes in Coastline, Climate, and Human Settlement. Dordrecht: Springer, 2007, pp 775–796.
Nichols J : The origin of the Chechen and Ingush: A study in alpine linguistic geography. Anthropological Linguistics 2005; 46: 129–155.
Nichols J : The Nakh-Daghestanian consonant correspondences: in: Tuite K, Holisky DA (eds): Current trends in Caucasian, East European, and Inner Asian linguistics: Papers in honor of Howard I. Aronson. Amsterdam: Benjamins, 2003, pp 207–251.
Embleton SM : Statistics in Historical Linguistics. Bochum: Brockmeyer, 1986.
Bulaeva KB, Dubinin NP, Shamov IA, Isaichev SA, Pavlova TA : [Population genetics of Dagestan highlanders]. Genetika 1985; 21: 1749–1758.
Bulayev OA, Gurgenova FR, Guseinova UM, Bulaeva KB : [Mapping genes of major recurrent depression in genetic isolates]. Zh Nevrol Psikhiatr Im S S Korsakova 2011; 111: 62–69.
Bulaeva KB, Pavlova TA, Kurbanov RM, Bulaev OA : [Mapping genes of complex diseases in genetic isolates of Dagestan]. Genetika 2002; 38: 1539–1548.
Bulayeva KB : Overview of genetic-epidemiological studies in ethnically and demographically diverse isolates of Dagestan, Northern Caucasus, Russia. Croat Med J 2006b; 47: 641–648.
Bulayeva KB, Glatt SJ, Bulayev OA, Pavlova TA, Tsuang MT : Genome-wide linkage scan of schizophrenia: a cross-isolate study. Genomics 2007; 89: 167–177.
Bulayeva KB, Leal SM, Pavlova TA et al: The ascertainment of multiplex schizophrenia pedigrees from Daghestan genetic isolates (Northern Caucasus, Russia). Psychiatr Genet 2000; 10: 67–72.
Guseinov GG, Shamov IA, Bulaeva KB : [A genetic epidemiological study of arterial hypertension in an isolated mountain area of Dagestan]. Ter Arkh 1996; 68: 64–66.
Illarioshkin SN, Ivanova-Smolenskaia IA, Dimborskaia SA et al: [Mapping of the gene for autosomal-recessive progressive muscular dystrophy in an isolate from a highland region of Dagestan to chromosome 2-13]. Genetika 1997; 33: 1551–1558.
Bulaeva KB, Davudov OM, Pavlova TA, Kurbanov RM, Bulaev OA, Harpending H : [Genetic subdivision of Dagestan ethnic populations]. Genetika 2003b; 39: 83–92.
Balanovsky O, Dibirova K, Dybo A et al: Parallel evolution of genes and languages in the Caucasus region. Mol Biol Evol 2011; 28: 2905–2920.
Bulayeva K, Jorde LB, Ostler C, Watkins S, Bulayev O, Harpending H : Genetics and population history of Caucasus populations. Hum Biol 2003a; 75: 837–853.
Caciagli L, Bulayeva K, Bulayev O et al: The key role of patrilineal inheritance in shaping the genetic variation of Dagestan highlanders. J Hum Genet 2009; 54: 689–694.
Marchani EE, Watkins WS, Bulayeva K, Harpending HC, Jorde LB : Culture creates genetic structure in the Caucasus: autosomal, mitochondrial, and Y-chromosomal variation in Daghestan. BMC Genet 2008; 9: 47.
Nasidze I, Ling EY, Quinque D et al: Mitochondrial DNA and Y-chromosome variation in the caucasus. Ann Hum Genet 2004; 68: 205–221.
Nasidze I, Sarkisian T, Kerimov A, Stoneking M : Testing hypotheses of language replacement in the Caucasus: evidence from the Y-chromosome. Hum Genet 2003; 112: 255–261.
Tofanelli S, Ferri G, Bulayeva K et al: J1-M267 Y lineage marks climate-driven pre-historical human displacements. Eur J Hum Genet 2009; 17: 1520–1524.
Yunusbayev B, Metspalu M, Jarve M et al: The Caucasus as an asymmetric semipermeable barrier to ancient human migrations. Mol Biol Evol 2011; 29: 359–365.
Hammer MF, Karafet TM, Redd AJ et al: Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 2001; 18: 1189–1203.
Karafet TM, Zegura SL, Posukh O et al: Ancestral Asian source(s) of new world Y-chromosome founder haplotypes. Am J Hum Genet 1999; 64: 817–831.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Excoffier L, Lischer HE : Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10: 564–567.
Behar DM, Yunusbayev B, Metspalu M et al: The genome-wide structure of the Jewish people. Nature 2010; 466: 238–242.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Rosenberg NA, Pritchard JK, Weber JL et al: Genetic structure of human populations. Science 2002; 298: 2381–2385.
Team RDC. R A language and environment for statistical computing. Vienna, Austria: R Foundation for Statisrical Computing, 2010.
Jakobsson M, Scholz SW, Scheet P et al: Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008; 451: 998–1003.
McEvoy BP, Powell JE, Goddard ME, Visscher PM : Human population dispersal "Out of Africa" estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res 2011; 21: 821–829.
Tenesa A, Navarro P, Hayes BJ et al: Recent human effective population size estimated from linkage disequilibrium. Genome Res 2007; 17: 520–526.
Hayes BJ, Visscher PM, McPartlan HC, Goddard ME : Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res 2003; 13: 635–643.
Gibson J, Morton NE, Collins A : Extended tracts of homozygosity in outbred human populations. Hum Mol Genet 2006; 15: 789–795.
Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF : Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010; 5: e13996.
Jakkula E, Rehnstrom K, Varilo T et al: The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet 2008; 83: 787–794.
Ripke S, Neale B, Corvin A et al: Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Aglarov MA : [Rural society in Mountainous Daghestan in XVII - the beginning XIX centuary]. Moscow: Nauka, 1988.
Bulayeva KB, Davudov OM, Pavlova TA, Kurbanov RM, Bulaev OA, Harpending H : [Genetic subdivision of Dagestan ethnic populations]. Genetika 2003b; 39: 83–92.
Bulayeva KB, Jorde L, Watkins S et al: Ethnogenomic diversity of Caucasus, Daghestan. Am J Hum Biol 2006a; 18: 610–620.
Howrigan DP, Simonson MA, Keller MC : Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics 2011; 12: 460.
Roy-Gagnon MH, Moreau C, Bherer C et al: Genomic and genealogical investigation of the French Canadian founder population structure. Hum Genet 2011; 129: 521–531.
Teo SM, Ku CS, Salim A, Naidoo N, Chia KS, Pawitan Y : Regions of homozygosity in three Southeast Asian populations. J Hum Genet 2012; 57: 101–108.
Bozhkova VP, Khashaev ZH, Umanskaya TM : [Frequence and the mutation spectrum of GJB2 related hearing loss in children of Dagestan as compared with the Central European part of Russia]. Biofizika 2010; 55: 514–525.
Bulayev OA, Pavlova TA, Bulayeva KB : [The effect of inbreeding on accumulation of complex diseases in genetic isolates]. Genetika 2009; 45: 1096–1104.
Bulayeva K, Lencz T, Glatt S et al: [Mapping genes related to early onset major depressive disorder in dagestan genetic isolates]. Turk Psikiyatri Derg 2011; 23: 161–170.
Bulayeva KB, Leal SM, Pavlova TA et al: Mapping genes of complex psychiatric diseases in Daghestan genetic isolates. Am J Med Genet B Neuropsychiatr Genet 2005; 132B: 76–84.
Bulayeva KB, Pavlova TA, Kurbanov RM, Bulaev OA : [Mapping genes of complex diseases in genetic isolates of Dagestan]. Genetika 2002; 38: 1539–1548.
Bulayeva KB, Pavlova TA, Kurbanov RM, Leal S, Bulayev OA : [Genetic and epidemiological studies in Dagestan highland isolates]. Genetika 2003c; 39: 413–422.
Neel JV : Minority populations as genetic isolates: The interpretation of inbreeding results in: Bittles AH, Roberts DF (eds): Minority Populations: Genetics, Demography and Health. London: Macmillan, 1992, pp 1–13.
Jorde LB : Linkage disequilibrium and the search for complex disease genes. Genome Res 2000; 10: 1435–1444.
Lescai F, Franceschi C : The impact of phenocopy on the genetic analysis of complex traits. PLoS ONE 2010; 5: e11876.
Zuk O, Schaffner SF, Samocha K et al: Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 2014; 111: E455–E464.
Hatzikotoulas K, Gilly A, Zeggini E : Using population isolates in genetic association studies. Brief Funct Genomics 2014; 13: 371–377.
Glodzik D, Navarro P, Vitart V et al: Inference of identity by descent in population isolates and optimal sequencing studies. Eur J Hum Genet 2013; 21: 1140–1145.
Acknowledgements
We thank all Daghestanian people participating in this study, August Woerner and Brian Hallmark for bioinformatics support. This work was supported by grants from the National Science Foundation (BSC 1025266 to MFH and TMK and BSC0742328 to TMK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Karafet, T., Bulayeva, K., Bulayev, O. et al. Extensive genome-wide autozygosity in the population isolates of Daghestan. Eur J Hum Genet 23, 1405–1412 (2015). https://doi.org/10.1038/ejhg.2014.299
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.299
This article is cited by
-
Characterization of Danube Swabian population samples on a high-resolution genome-wide basis
BMC Genomics (2023)
-
Runs of homozygosity: windows into population history and trait architecture
Nature Reviews Genetics (2018)
-
Weighted likelihood inference of genomic autozygosity patterns in dense genotype data
BMC Genomics (2017)
-
Overcoming the dichotomy between open and isolated populations using genomic data from a large European dataset
Scientific Reports (2017)
-
Between Lake Baikal and the Baltic Sea: genomic history of the gateway to Europe
BMC Genetics (2017)
