
Abstract
Variants in ACTA1, which encodes α-skeletal actin, cause several congenital myopathies, most commonly nemaline myopathy. Autosomal recessive variants comprise approximately 10% of ACTA1 myopathy. All recessive variants reported to date have resulted in loss of skeletal α-actin expression from muscle and severe weakness from birth. Targeted next-generation sequencing in two brothers with congenital muscular dystrophy with rigid spine revealed homozygous missense variants in ACTA1. Skeletal α-actin expression was preserved in these patients. This report expands the clinical and histological phenotype of ACTA1 disease to include congenital muscular dystrophy with rigid spine and dystrophic features on muscle biopsy. This represents a new class of recessive ACTA1 variants, which do not abolish protein expression.
Similar content being viewed by others


Introduction
Variants in the skeletal muscle α-actin gene (ACTA1) are a common cause of congenital myopathy with over 200 different variants now reported.1 By far, the most frequent histological pattern associated is autosomal dominant nemaline myopathy; less common patterns include actin-accumulation myopathy, intranuclear rod myopathy and congenital fiber-type disproportion.1 Approximately 10% of ACTA1-myopathy patients have autosomal recessive disease, and until now all such patients have had nemaline myopathy and a severe congenital phenotype associated with loss of α-skeletal actin expression.1 We report a novel homozygous recessive missense variant (c.460G>C, P.(Val154Leu)) in two brothers with infantile-onset congenital muscular dystrophy with rigid spine, thus expanding the histological and clinical phenotypes associated with ACTA1 variants and establishing a new class of ACTA1 variants – recessive variants associated with expression of mutant actin and mild-to-moderate disease severity.
Patients and methods
Patients
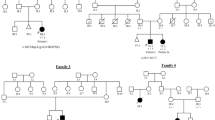
We enrolled a large cohort of patients with limb-girdle weakness who lacked a genetic diagnosis in a whole-exome sequencing study, conducted with approval from the Human Research Ethics Committee of the Sydney Children’s Hospitals Network (10/CHW/45).
Genetic analysis
DNA was extracted from blood leukocytes of Patient 1 using standard techniques. A QIAGEN (Melbourne, VIC, Australia) QIAamp DNA FFPE Tissue kit was used to extract DNA from formalin-fixed paraffin-embedded skeletal muscle of Patient 2.
Targeted next-generation sequencing of 45 neuromuscular disease genes was performed by emulsion PCR (RainDance, Billerica, MA, USA) of 1840 amplimers followed by sequence analysis (Illumina HiScan SQ). Data interpretation were performed using NextGene software. Confirmatory Sanger sequencing was carried out by the Australian Genome Research Facility, Sydney. The sequence was aligned on Sequencer 4.9 (Ann Arbor, MI, USA) with the reference sequence from NCBI, NM_001100.3. The variant is submitted to the Leiden Muscular Dystrophy ACTA1 Mutation Database (www.LOVD.nl/ACTA1 (database ID ACTA1_00245)). Molecular modeling of the α-skeletal actin monomer was performed using DeepView/Swiss-Pdb Viewer (v 4.1.0, Basel, Switzerland), http://www.expasy.org/spdbv/2 from the crystal structure solved by Kabsch et al.3
Muscle biopsy analysis
Muscle histology slides were stained using standard procedures. Frozen muscle biopsy sections were stained with antibodies to α-skeletal actin, cardiac actin and neonatal myosin. Western blot analysis was performed using antibodies to α-skeletal actin and cardiac actin. Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control. See Supplementary methods for further details.
Results
Clinical features
A Sri Lankan couple with possible distant consanguinity had two affected sons and four healthy children. The affected boys’ prenatal and perinatal periods were uncomplicated and both walked at around 1 year of age. Neck weakness was noted in infancy in Patient 1 and by age 2 years, both were unable to run and fell frequently. Patient 1 had mild, stable, generalized weakness during childhood that progressed slowly from late teenage years. A mild thoracic kyphoscoliosis developed in adolescence. In early adulthood he developed mild dysphagia and mild restrictive lung disease. At age 25 years, forced vital lung capacity measured 44% of predicted. Nocturnal ventilation was not required. At age 34 years, he walked short distances and had a rigid spine (Figure 1a), generalized muscle wasting and a body mass index of 15. There was mild facial weakness, restricted jaw opening, scapular winging and generalized weakness in all limbs, worse in proximal muscles.

Clinical and radiological features associated with ACTA1 variant Val154Leu. (a) Patient 1 at age 34 years demonstrating maximum forward flexion of the spine and neck. (b and c) Patient 1 muscle MRI of the left mid thigh (b) showing prominent fat infiltration in the rectus femoris and muscles of the posterior compartment (arrows), and left lower leg (c) showing particular involvement of the soleus (arrow).
At age 5 years, Patient 2 was ambulant but used a wheelchair for longer distances. He had generalized weakness, more severe in proximal limb muscles with a positive Gowers’ sign. He suffered recurrent pneumonia and died during one episode at age 6 years. Neither boy had ptosis, ophthalmoplegia, nor limb contractures.
Creatine kinase measures varied between 222and 1930 U/l. The electromyogram was myopathic in both siblings. Nerve conduction studies and echocardiograms were normal. Muscle MRI in Patient 1 at 34 years showed widespread muscle atrophy and fat infiltration (Figure 1b and c). At the mid thigh there was relative sparing of the vastus lateralis, vastus medialis, gracilis and sartorius muscles, while the posterior compartment and rectus femoris were more severely affected. In the lower leg there was mild to moderate generalized fatty infiltration, especially of soleus.
Genetic analysis
A missense variant (c.460G>C, p.(Val154Leu)) in ACTA1 was identified in Patient 1. Sanger sequencing confirmed that both brothers were homozygous for the variant, with both parents heterozygous for the change. All unaffected siblings were either heterozygous or homozygous for the normal allele. A previously unreported, heterozygous missense variant in SEPN1 (NM_020451.2: c.1162A>G, p.(Ser388Gly) (Leiden Muscular Dystrophy SEPN1 Mutation Database (www.LOVD.nl/SEPN1 (database ID SEPN1_00115)) was also identified in Patient 1 but a second SEPN1 variant was not found with Sanger sequencing or on a custom screen for exon deletions or duplications. No variants were found in other genes previously associated with a rigid spine phenotype (CAPN3, COL6A1, COL6A2, COL6A3 or LMNA).
Molecular modeling showed that the Val154Leu variant alters a residue in subdomain 3 situated near the ATP-binding pocket and the hinge region (Figure 2h). The hinge region is responsible for relative movement of the domains in respect to each other, allowing molecules, including ATP, to bind in the clefts. As flexibility of the relative domains is essential for polymerization of the actin filament, the Val154Leu variant may influence polymerization by influencing the hinge region. The relative movement of the two domains may be changed such that ATP binds less efficiently.

Histological features associated with ACTA1 variant Val154Leu. (a) Electron microscopy of Patient 1’s quadriceps muscle biopsy shows accumulated Z-band material. (b) Hematoxylin and eosin staining of Patient 1 quadriceps muscle shows multiple internalized nuclei (i), a regenerating fiber (ii) and a likely split fiber (iii). (c) Electron microscopy of Patient 1’s quadriceps muscle biopsy shows streaming and accumulation (i) of Z-band material. Occasional small rods are also present (arrows). (d) COX SDH staining of Patient 1 quadriceps muscle biopsy demonstrating central lucencies and ‘moth-eaten’ areas suggestive of cores. (e) One percent of myofibers in Patient 1 was positive for neonatal myosin, supporting the presence of occasional regenerating fibers. (f) Immunohistochemistry for skeletal α-actin shows normal expression in all myofibers in muscle from Patient 1 at 17 years of age compared with adult control (g). (h) Molecular modeling of α-skeletal actin monomer in DeepView/Swiss-Pdb Viewer (v 4.1.0, http://www.expasy.org/spdbv/) from the crystal structure solved by Kabsch et al.3 Valine154 is shown in yellow space fill, on actin depicted as a black ribbon structure. The two polypeptide chains making up the hinge region are shown in blue. (i) Immunohistochemistry of Patient 1 muscle demonstrates the presence of cardiac α-skeletal actin in 20% of myofibers of variable size and staining intensity, compared with adult control skeletal muscle (j). (k and l) Western blot analysis of Patient 1 muscle with glyceraldehyde 3-phosphate dehydrogenase shown as a loading control. (k) Normal levels of α-skeletal actin are present in Patient 1 muscle. Lanes 1 and 3 show adult control skeletal muscle, lane 2 contains patient skeletal muscle. (l) Cardiac actin is markedly upregulated in Patient 1 skeletal muscle compared with normal controls. Lane 1 contains baboon cardiac muscle, lanes 2 and 4 contain control adult skeletal muscle and lane 3 contains muscle from Patient 1. The full colour version of this figure is available at European Journal of Human Genetics online.
Muscle biopsy analysis
A quadriceps muscle biopsy of Patient 1 at age 17 years 7 months showed increased fiber size variation, numerous internalized nuclei, fiber splitting, occasional degenerating and regenerating fibers (Figure 2b). Thirty-four percent of myofibers had at least one internal or central nucleus and 13% had two or more. The nicotinamide adenine dinucleotide tetrazolium reductase (NADH), cytochrome oxidase (COX) and succinic dehydrogenase (SDH) stains showed central lucencies and a ‘moth-eaten appearance’, suggestive of cores (Figure 2d). Similar findings, with prominent fatty infiltration, were present in a quadriceps biopsy from Patient 2 at age 5 years.
In both patients, electron microscopy (EM) showed smeared and accumulated Z-band material that typically spanned 1–2 sarcomeres in length, but extended across up to half the width of the fiber. The degree of accumulated Z-band material is unusual for minicores (Figure 2a). A single more typical minicore, lacking the accumulated Z-band material, was seen in the biopsy from Patient 2. Occasional small nemaline rods were present (Figure 2c). These were too small and infrequent to be seen on light microscopy on Gomori trichrome, or with staining for cardiac and α-skeletal actin, or α-actinin.
Immunohistochemical staining for neonatal myosin on Patient 1 muscle showed 1% positive fibers (Figure 2e), consistent with the presence of occasional regenerating fibers. Cardiac α-actin was present in 20% of myofibers of variable size and fiber type, and with variable staining intensity (Figure 2i and j). Staining for α-skeletal actin showed normal levels of expression in all myofibers, indicating that mutant p.(Val154Leu) actin is expressed and is the predominant actin isoform in most myofibers (Figure 2f and g).
Western blot analysis of Patient 1 muscle showed normal levels of α-skeletal actin (Figure 2k). Cardiac actin was markedly upregulated compared with normal controls (Figure 2l).
Discussion
The ACTA1 gene is highly conserved throughout evolution and polymorphisms that alter the protein sequence are extremely rare in the general population, indicating that most changes to the actin polypeptide sequence adversely affect protein function.4 All previously described patients with recessive ACTA1 disease (∼10% of ACTA1 patients) have had functional ‘null’ variants and make no α-skeletal actin.1, 4, 5 All have had severe congenital nemaline myopathy and survival after birth is only possible due to persistence of cardiac α-actin in the skeletal muscle of these children.5 We describe the first recessive ACTA1 variant associated with expression of α-skeletal actin in skeletal muscle and survival well into adulthood. A limitation of this study is that we have not performed a comprehensive screen for other genetic variants that may influence the patient’s phenotype. Nevertheless, the ACTA1 recessive variants segregate with disease in the family and are predicted to be pathogenic by molecular modeling. As in other recessive ACTA1 myopathies, cardiac actin was upregulated in patient muscle, but only in a subset of myofibers. Occasional cardiac actin-positive fibers may be regenerating fibers in which fetal sarcomeric isoforms, including cardiac actin and neonatal myosin, are expressed, but this explanation is unlikely to account for most cardiac actin-positive myofibers. We suspect cardiac actin is upregulated as a specific response to abnormalities in α-skeletal actin, as cardiac actin expression is unusual in congenital myopathies or dystrophies after the neonatal period, except in the context of myofiber regeneration.
The finding of mild dystrophic changes on muscle biopsy has not been previously reported in association with variants in ACTA1 and expands the histological phenotype for ACTA1-related myopathies. Another notable histological feature was the presence of prominent internalized nuclei. Extensive Z-band streaming and the presence of occasional rods on EM may also provide a clue to the genetic diagnosis.
On clinical examination, spinal rigidity was a notable feature and the mild to moderately raised CK levels in both probands, together with the muscle biopsy findings led to a clinical diagnosis of congenital muscular dystrophy with spinal rigidity. Congenital muscular dystrophy with a rigid spine and variably elevated CK has been previously described in association with variants in SEPN1, LMNA and collagen VI genes, and we screened these genes for variants.6 The lack of limb contractures in Patient 1 by adulthood is also atypical for LMNA- and COLVI-related myopathies, and almost all SEPN1-related myopathy patients develop nocturnal hypoventilation by this age. These clinical features and a different pattern of lower limb muscle involvement in our family may be useful clinical clues to identify additional patients with ACTA1-CMD.
This report extends the phenotypic spectrum of ACTA1 myopathies to include congenital muscular dystrophy associated with rigid spine. Until recently, genetic testing has generally been limited to the analysis of disease genes known to be associated with the patient’s phenotype and expense has been a major barrier to broad genetic screens. Advances in sequencing technologies now mean that testing of all known myopathy genes is widely available for patient diagnosis and there are many examples of when such testing has revealed unexpected genotype–phenotype associations.7 Even though ACTA1 was one of the first known congenital myopathy genes,8 and ACTA1 diagnostic sequencing has been available for over 14 years, the family we report raises the possibility that variants in ACTA1 may be responsible for a much wider spectrum of disease than is currently appreciated.
References
Nowak KJ, Ravenscroft G, Laing NG : Skeletal muscle α-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathol 2012; 125: 19–32.
Guex N, Peitsch MC : SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 1997; 18: 2714–2723.
Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC : Atomic structure of the actin: DNase I complex. Nature 1990; 347: 37–44.
Laing NG, Dye DE, Wallgren-Pettersson C et al: Mutations and polymorphisms of the skeletal muscle α-actin gene (ACTA1). Hum Mutat 2009; 30: 1267–1277.
Nowak KJ, Sewry CA, Navarro C et al: Nemaline myopathy caused by absence of α-skeletal muscle actin. Ann Neurol 2007; 61: 175–184.
Mercuri E, Clements E, Offiah A et al: Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010; 67: 201–208.
Ceyhan-Birsoy O, Agrawal PB, Hidalgo C et al: Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 2013; 81: 1205–1214.
Nowak KJ, Wattanasirichaigoon D, Goebel HH et al: Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 1999; 23: 208–212.
Acknowledgements
This study was supported by National Health and Medical Research Council (Australia) grants 1022707, 1031893 & 1026933 (KNN, NFC), and 1056285 (GLO); National Institutes of Health (USA) 3R01 NS29525 (EPH); and The Australian Postgraduate Award, The University of Sydney (HAB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
GL O’Grady: Drafting/revising manuscript for content, analysis and interpretation of data. HA Best: Drafting/revising manuscript for content, analysis and interpretation of data. EC Oates: Drafting/revising manuscript for content, data generation, data interpretation. S Kaur: Data generation, data interpretation, manuscript revision. A Charlton: Revising manuscript for content, analysis and interpretation of data. S Brammah: Revising manuscript for content, analysis and interpretation of data. E Hoffman: Revising manuscript for content, study concept, analysis and interpretation of data. A Kesari: Experimental design, data generation, data interpretation, revising manuscript. J Punetha: Experimental design, data generation, data interpretation, revising manuscript. KN North: Drafting/revising manuscript for content, study concept and design, analysis and interpretation of data, study supervision, obtaining funding. Biljana Ilkovski: Drafting/revising manuscript for content, analysis and interpretation of data. NF Clarke: Drafting/revising manuscript for content, study concept and design, analysis and interpretation of data, study supervision, obtaining funding.
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
O'Grady, G., Best, H., Oates, E. et al. Recessive ACTA1 variant causes congenital muscular dystrophy with rigid spine. Eur J Hum Genet 23, 883–886 (2015). https://doi.org/10.1038/ejhg.2014.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.169
This article is cited by
-
Nemaline myopathies: a current view
Journal of Muscle Research and Cell Motility (2019)
-
Muscular MRI-based algorithm to differentiate inherited myopathies presenting with spinal rigidity
European Radiology (2018)
-
Clinical utility gene card for: Nemaline myopathy – update 2015
European Journal of Human Genetics (2015)
-
Zebrafish models for nemaline myopathy reveal a spectrum of nemaline bodies contributing to reduced muscle function
Acta Neuropathologica (2015)
