
Abstract
PRPS1 codes for the enzyme phosphoribosyl pyrophosphate synthetase-1 (PRS-1). The spectrum of PRPS1-related disorders associated with reduced activity includes Arts syndrome, Charcot–Marie–Tooth disease-5 (CMTX5) and X-linked non-syndromic sensorineural deafness (DFN2). We describe a novel phenotype associated with decreased PRS-1 function in two affected male siblings. Using whole exome and Sanger sequencing techniques, we identified a novel missense mutation in PRPS1. The clinical phenotype in our patients is characterized by high prenatal maternal α-fetoprotein, intrauterine growth restriction, dysmorphic facial features, severe intellectual disability and spastic quadraparesis. Additional phenotypic features include macular coloboma-like lesions with retinal dystrophy, severe short stature and diabetes insipidus. Exome sequencing of the two affected male siblings identified a shared putative pathogenic mutation c.586C>T p.(Arg196Trp) in the PRPS1 gene that was maternally inherited. Follow-up testing showed normal levels of hypoxanthine in urine samples and uric acid levels in blood serum. The PRS activity was significantly reduced in erythrocytes of the two patients. Nucleotide analysis in erythrocytes revealed abnormally low guanosine triphosphate and guanosine diphosphate. This presentation is the most severe form of PRPS1-deficiency syndrome described to date and expands the spectrum of PRPS1-related disorders.
Similar content being viewed by others


Introduction
PRPS1 (Xq22.3) codes for the enzyme phosphoribosylpyrophosphate synthetase-1 (PRS-1), which catalyzes the first step of nucleotide synthesis necessary for the de novo and salvage pathways of purine and pyrimidine biosynthesis.1 Mutations in PRPS1 may result in gain or loss of function of the protein, and the clinical phenotype depends on the effect of the mutation in different cell types. To date, the spectrum of PRPS1-related disorders associated with loss-of-function mutations includes Arts syndrome2 (MIM 301835), Charcot–Marie–Tooth disease-53 (CMTX5, MIM 311070) and X-linked non-syndromic sensorineural deafness4 (DFN2, MIM 304500). Other two phenotypes associated with gain-of-function mutations include PRPS-related gout (MIM 300661) and phosphoribosylpyrophosphate synthetase superactivity5 (MIM 300661).
PRPS1 mutations are associated with considerable phenotypic heterogeneity. Patients with CMTX5 present with ataxia, hypotonia, loss of deep tendon reflexes, hearing impairment and optic atrophy, whereas DNF2 is characterized by isolated hearing impairment. For both disorders, intellectual function is normal. Arts syndrome was first described in a family with 12 affected males, 11 of whom died before 5 years of age. These patients presented with intellectual disability, early-onset hypotonia, ataxia, delayed motor development, hearing impairment, optic atrophy and susceptibility to infections.2 De Brouwer and co-workers6 reported the original Dutch family described by Arts et al,2 in addition to two additional Australian male patients. They all had undetectable hypoxanthine in urine and reduced uric acid levels in the serum. In vitro, PRS-1 activity was reduced in erythrocytes and fibroblasts, and loss-of-function missense mutations in PRPS1 were identified in these patients.
Here, we describe a novel phenotype associated with decreased PRS-1 function, characterized by prenatal high maternal α-fetoprotein (MS-AFP) and intrauterine growth restriction, and postnatal dysmorphic facial features, severe intellectual disability and spastic quadraparesis. In addition, these patients developed Leber’s congenital amaurosis, diabetes insipidus and short stature.
Materials and methods
Patients
Patients were recruited from the Genetics Clinic at The Hospital for Sick Children (Toronto, ON, Canada). Consent for exome sequencing and confirmatory molecular and biochemical studies were obtained following the hospital research ethics board policies. Samples were obtained from the affected individuals, their clinically unaffected brother and additional relatives at risk of being carriers for the mutation identified. The clinical phenotype was assessed as per the standard of care by the respective subspecialists at The Hospital for Sick Children.
Exome sequencing methods
Paired-end (75+35 nt) exome sequencing was performed using Life Technologies SOLiD 5500xL (Life Technologies, Foster City, CA, USA) sequencing platform after target enrichment with the Agilent SureSelect 50 Mb Human All Exon Capture Kit V3 (Agilent Technologies, Santa Clara, CA, USA). Sequenced reads were mapped to the reference human genome (UCSC hg19) using BFAST (http://bfast.sourceforge.net).
Variant calls were made using GATK version1.0.5506 and recommended parameters (Depristo, Nat Gen, PMID: 21478889). Base quality score recalibration optimized to SOLID 5500xl was used to remove the reference bias. To differentiate common polymorphisms from novel variants, identified single-nucleotide variants (SNVs) and insertion/deletions (indels) were compared with public variant databases (dbSNP137; Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/) and the 1000 genomes project7). A high confident SNV was defined as having a quality by depth (QD) ≥5.0, a strand bias (SB) ≤−0.01 and GATK filter of ‘pass’ according to GATK recommended cutoffs. A high confidence indel was defined as having a QD ≥10.0 and an SB ≤−0.01 according to GATK recommended cutoffs.
Phosphoribosyl pyrophosphates assay
Erythrocytes were isolated from EDTA blood (5 ml) by centrifugation at 800 g for 5 min at room temperature (RT). The plasma and leukocyte fractions were discarded and the pellet containing the erythrocytes was supplemented with 0.9% (w/v) NaCl to the original volume of the blood sample (5 ml) and centrifuged (800 g, 20 min, RT). The top part of the erythrocyte pellet was removed and the erythrocytes were washed once more with 0.9% (w/v) NaCl. The final erythrocyte pellet was diluted 1:1 with 0.9% (w/v) NaCl and exactly 100 μl was snap frozen in liquid nitrogen and stored at −80 °C.
Frozen-packed erythrocytes (100 μl) were diluted with 400 μl of 0.9% (w/v) NaCl and concentrated on a Amicon Ultra Ultracel 10K Membrane filter (Millipore, Billerica, MA, USA) by centrifugation (14 000 g at 4 °C for 60 min). The protein fraction was diluted to a final volume of 500 μl with 0.9% (w/v) NaCl and concentrated again by centrifugation. This procedure was repeated once more and the protein concentration determined with a copper-reduction method using bicinchonic acid as described by Smith et al.8
The activity of phosphoribosyl pyrophosphate (PRPPs) was determined in a reaction mixture (110 μl) containing an aliquot of cell sample (0.1–0.5 mg per assay), 32 mM sodium phosphate, 1 mM dithiothreitol, 4.5 mM MgCl2, 1 mM ATP, 100 μ M ribose-5-phosphate and 50 mM TRIS/MOPS (pH7.4). Separation of AMP, ADP and ATP was performed using a gradient from 100% buffer A (7.5 mM sodium phosphate) to 70% buffer B (0.75 mM sodium phosphate (pH 4.55)) in 25 min, at a flow rate of 1.0 ml/min by high-performance liquid chromatography (HPLC) on a ion exchange column (Whatman Partisphere SAX 125 × 4.6 mm2 (Whatman, Clifton, NJ, USA), 5 μm particle size) and a guard column (Whatman Partisphere AX 10 × 2.5 mm2, 5 μm particle size) with online UV detection at 254 nm. Protein concentrations were determined with a copper-reduction method using bicinchonic acid, essentially as described by Smith et al.8
Nucleotide analysis
Erythrocytes were isolated, essentially as described above with minor modifications: phosphate-buffered saline was used instead of 0.9% (w/v) NaCl. Ribonucleotides were extracted by adding 500 μl of ice-cold 0.8 M perchloric acid to 100 μl of packed erythrocytes. After 10 min on ice, the suspension was centrifuged at 11 000 g at 4 °C for 15 min. The supernatant (150 μl) was removed and neutralized with approximately 7.5 μl of 5.0 M K2CO3. After centrifugation (11 000 g at 4 °C for 5 min), the supernatant was saved and used for HPLC analysis. The dry pellet containing total protein was dissolved in 500 μl of 0.2 M NaOH and the protein content was measured with a copper-reduction method using bicinchonic acid, essentially as described by Smith et al.8
Nucleotide profiles were determined by ion-exchange HPLC, using a Whatman Partisphere SAX 4.6 × 125 mm2 column (5 μm particles) and a Whatman 10 × 2.5 mm2 AX guard column. The buffers used were 9 mM NH4H2PO4, pH 3.5 (buffer A) and 325 mM NH4H2PO4, 500 mM KCl, pH 4.4 (buffer B). Nucleotides were eluted with a gradient from 100% buffer A to 90% buffer B in a total run time of 60 min, at a flow rate of 1 ml/min.9
Results
Clinical findings
Patient 1
A male proband was born to a 29-year-old mother (gravida 1, para 0) and 33-year-old father of Caucasian descent. The parents were healthy, non-consanguineous and their family history was unremarkable. Routine pregnancy screen at 16 weeks gestational age (GA) showed high MS-AFP. Fetal ultrasound at 18 weeks GA showed normal fetal anatomy and placenta, and amniocentesis showed normal male karyotype. Serial fetal ultrasounds at 21 and 25 weeks GA were normal. The mother was healthy throughout pregnancy. The child was delivered at 31 weeks GA by emergency Cesarean section, following an ultrasound that showed significant intrauterine growth retardation and low biophysical profile. His APGAR scores were 0 and 1 at 1 and 5 min, respectively. The birth weight was 911 g (−3 standard deviation (SD)). The head circumference was 23.5 cm (10th centile). The baby required respiratory support and endotracheal intubation for 2 weeks. He remained in the neonatal intensive care unit (NICU) for 6 weeks, where he developed jaundice requiring phototherapy. There was no history of significant infections, seizures or hypoglycemia. He was transitioned from nasogastric tube feeding to bottle feeding at the age of 3 weeks postdelivery.
The patient at the age of 6 years showed significant developmental delay. He had profound intellectual disability and did not acquire developmental milestones beyond the age of 2–3 months. The boy showed no social smile nor did he recognize his parents or turn his head to sound. At 3 years of age, he was diagnosed with diabetes insipidus that responded well to desmopressin. He continued to have significant failure to thrive and G tube feeding was initiated at age 4 years. At 5 years of age, he was diagnosed with generalized tonic clonic seizures that were brief and infrequent and did not require antiepileptic medication. His weight and height were at −3 and −4 SD, respectively. The head circumference was between the 3rd and 5th centile. He had dysmorphic features including dolichocephalic cranium, deep-set eyes, hypertelorism, hypoplastic nares and bilateral low-set ears (Figure 1). Neurologically, he had significant head lag and truncal hypotonia associated with increased tone of the upper and lower limbs with brisk deep tendon reflexes and clonus. There were no extrapyramidal movements and he did not develop signs suggestive of normal vision. There was no history of recurrent infections.

Clinical findings. Patients 1 (a and c) and 2 (b and d) presented with strikingly similar dysmorphic features and neurological phenotype. Characteristic facial features included dolichocephalic skull, hypertelorism, deep-set eyes, hypoplastic nares and low-set ears. Additionally, the patients developed truncal hypotonia and axial hypertonia.
Eye examination showed light perception, exotropia and a sluggish pupillary response to light. Fundus examination showed changes compatible with a retinal dystrophy, including attenuated vessels, pigmentary changes, peripheral vascular anomalies, a central chorioretinal atrophy (‘macular coloboma’) and bilateral pale optic discs. No nystagmus was observed. Visual-evoked potentials (VEP) revealed bilateral absent cortical response.
Array comparative genomic hybridization (Agilent, Oligo Array- 44K) identified a normal male chromosome complement with no microdeletions or microduplications (46, XY). DNA analysis by sequencing ARX, PLP1, CJA12 and MECP2 showed no detectable mutations. Extensive metabolic investigations including plasma amino acids, urine organic acids, blood lactate, ammonia, 7-dehydrocholesterol, cholesterol, transferring isoelectric focusing to screen for congenital disorders of glycosylation and very-long-chain fatty acids to exclude peroxisomal disorders were normal.
Patient 1 died suddenly in his sleep at the age of 10 years with no obvious cause and an autopsy was not performed. Between the birth of patient 1 and patient 2, the mother had two stillbirths, both of which were male fetuses. Pregnancy screen for both showed very high MS-AFP. An anatomical scan at 18 weeks was normal, and a follow-up of prenatal ultrasounds showed IUGR. The losses occurred at 23 and 24 weeks, respectively. The couple then had a healthy normal male, with a normal pregnancy and delivery (Supplementary Figure 1).
Patient 2
A male sibling was born to a 36-year-old mother and a 40-year-old father. The mother was healthy during pregnancy. For patient 2, routine pregnancy screen at 16 weeks GA also showed MS-AFP. Fetal ultrasound at 18 weeks GA showed normal fetal anatomy and placenta. At 27 weeks GA, a follow-up ultrasound showed intrauterine growth retardation. He was born by cesarean section at 29 weeks of gestation. His APGAR scores were 6 and 8 at 1 and 5 min, respectively. His birth weight was 740 g (−3 SD). He required respiratory support and endotracheal intubation for 1 day. He remained in the NICU for 8 weeks for weight gain, where he was bottle-fed. During his NICU admission, there was no history of significant infections, jaundice, seizures or hypoglycemia.
Patient 2 was also affected by profound developmental delay. At the age of 2 years, he was diagnosed with generalized tonic–clonic seizures, which were brief and infrequent and did not require antiepileptic medication. At 2.5 years of age, he presented with polyuria and polydypsia and he was diagnosed with diabetes insipidus. He responded well to desmopressin. He continued to have significant failure to thrive and G tube feeding was initiated at the age of 3 years. His weight was at −3 SD, height was −5 SD and head circumference was between the 3rd and 5th centile. He had dysmorphic features very similar to those of his affected brother, including dolichocephalic skull, deep-set eyes, hypertelorism, hypoplastic nares and bilateral low-set ears (Figure 1). He had significant head lag and truncal hypotonia. His limbs showed no spontaneous movement, increased tone and brisk deep tendon reflexes. He had bilateral positive Babinski sign.
Eye examination showed questionable light perception, exotropia and a sluggish pupillary response to light. Fundus examination showed similar changes compatible with a retinal dystrophy, including attenuated vessels, pigmentary changes, peripheral vascular anomalies, a central chorioretinal atrophy (‘macular coloboma’) and bilateral pale optic discs. Although no nystagmus was observed, this phenotype is suggestive of Leber’s congenital amaurosis. VEP showed bilateral absent cortical responses.
Electroencephalogram (EEG) at 4 years of age was abnormal and showed multiple spikes and polyspikes-and-wave activities over the posterior head region. Electrocardiogram showed no abnormal rhythm. Hearing test showed conductive hearing loss; his brain auditory-evoked potentials showed sensorineural hearing loss. Polysomnography at 7 years of age revealed significant central sleep apnea with oxygen desaturation, sleep fragmentation with no rapid eye movement (REM) sleep and decreased slow-wave sleep and abnormal EEG with frequent spikes. Extensive metabolic investigations including plasma amino acids, urine organic acids, blood lactate, ammonia, 7-dehydrocholesterol, cholesterol, congenital disorders of glycosylation screen and very long-chain fatty acids were normal. Additional investigations including complete blood count, thyroid function tests, growth hormone levels, serum cortisol levels, serum glucose and electrolytes, renal and liver function tests were within normal. There were no cardiac, gastrointestinal or genitourinary malformations and no history suggestive of immunodeficiency.
Neuroradiology
Patients 1 and 2 had two brain magnetic resonance imaging (MRI) investigations, both of which showed a thin corpus callosum with a tapered splenium and lack of isthmus (Figures 2 and 3). There was a boxy configuration to the calvarium with the suggestion of frontal bossing. Prominence of the anterior extra axial fluid spaces diminished over time. The lateral ventricles, particularly the frontal horns, were mildly prominent with mild flattening of the lateral walls of the frontal horns on initial imaging and at follow-up.

Neuroimaging results in patient 1. (a–d) MRI age 18 months: T1W sagittal image (a) shows a thin corpus callosum with the lack of isthmus and a tapered splenium (arrow). The optic chiasm is atrophied or hypoplastic. T1W axial image (b) demonstrates age-appropriate gyral development and mild delay in juxtacortical myelin maturation. T2W axial (c) and coronal (d) reveals prominence of the lateral ventricles, particularly anteriorly. There is flattening of the lateral walls of the frontal horns (arrow). Anterior pericerebral space is increased. (e–h) MRI age 6 years 10 months: T1W sagittal image (e) shows that corpus callosum remains thin, with a tapered splenium and with a lack of isthmus. The chiasm remains significantly small (arrow). MRS TE 144 (f) performed in the basal ganglia shows age-appropriate spectra. T2W axial images (g and h) reveal that myelin maturation is now age appropriate, although the white matter is thinned. The ventricles, in particular the anterior horns, remain prominent with flattening of the lateral walls (arrow).

Neuroimaging results in patient 2. (a–d) MRI age 8 months: T1W sagittal image (a) shows a thin corpus callosum with the lack of isthmus and a tapered splenium. The optic chiasm is atrophied or hypoplastic. T1W axial image (b) demonstrates age-appropriate gyral development. There is delay in myelin maturation, appropriate for 4–5 months of age. T2W axial (c) and coronal (d) reveals prominent lateral ventricles, particularly the frontal horns. The anterior pericerebral space is increased. (e–g) MRI at 5 years 2 months: T1W sagittal image (e) shows that corpus callosum remains thin, with a tapered splenium and with a lack of isthmus. The chiasm remains significantly small. The pituitary gland is small. Three-dimensional MRI surface reconstruction (f) reveals open mouth and mildly rotated pinna. T2W axial image (g) reveals age-appropriate myelin maturation. The white matter is thinned to a lesser degree than Child 1. The ventricles, particularly the frontal horns, remain prominent. T2W axial (h) view of the orbits demonstrates hypertelorism.
Myelin maturation, delayed in each, was normalized by the time of follow-up imaging. The white matter, however, was diminished in amount on follow-up imaging, although to a lesser degree in patient 2. Patient 2 also was noted to have a small pituitary gland on follow-up imaging. Ocular findings were similar in each, with definite hypertelorism, mild microphthalmia and significant atrophy or hypoplasia of the optic chiasm.
Brainstem and cerebellum were normal in configuration in each. Gyral development and MRS were always age appropriate. Neither child showed diffusion restriction on diffusion-weighted images or hemosiderin nor calcification on GRE sequences.
Exome sequencing
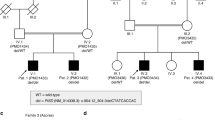
The affected male siblings and their mother were sequenced to a mean depth of coverage ranging from 33 to 41 × with at least 10 × coverage achieved in 69–70% of the targeted bases. Variant calling detected a total of 59 245 high-quality SNVs and indels among the three individuals. Of these, 3112 were coding variants with a frequency ≤5% in public databases (ESP, 1000 genomes project). Given the family history of multiple affected males (two born alive and two stillborns), we prioritized 19 variants on the X chromosome that were heterozygous in the mother for follow-up. A novel putative pathogenic mutation in the PRPS1 gene, located at chrX(GRCh37):g.106888462C>T, p.(Arg196Trp), c.586C>T (NM_002764.3), was confirmed by Sanger sequencing and segregated with the disorder (Figure 4). This arginine residue is highly conserved among vertebrates that have been sequenced for this region, and its replacement with tryptophan is predicted to be deleterious in silico (using the SIFT, PolyPhen2 and MutationTaster prediction programs). The data obtained in this study have been submitted to the Canadian controlled access database for phenotypic and genotypic information PhenomeCentral at https://phenomecentral.org.

Family pedigree and Sanger sequencing confirmation of the mutation and conservation of mutated residue across species.
Biochemical analysis
The patients had normal hypoxanthine concentrations in urine and uric acid levels in serum. Urine HPLC for purine and pyrimidine analysis in patient 2 showed reduced concentrations of adenosine, adenosylsuccinic acid, deoxyinosine, hypoxanthine and inosine, and elevation of the orotic acid (Table 1).
The PRS activity was significantly reduced in erythrocytes. The activity in erythrocytes as measured by HPLC was <0.002 nmol/mg/min in patient 1, < 0.0005 nmol/mg/min in patient 2, 1.51 nmol/mg/min in the mother, 1.23 nmol/mg/min in the maternal grandmother and 1.59 nmol/mg/min in the control sample. Nucleotide analysis in erythrocytes revealed abnormally low GTP, GDP, GTP+GDP and an abnormally increased (ATP+ADP)/(GTP+GDP) ratio (Table 2). These results suggest depletion in GTP that might affect central neurological functions for GTPase-dependent pathways such as dopamine synthesis.
Discussion
The spectrum of recognized disorders caused by disruption of PRS-1 function is broad and ranges from adult-onset gout to severe neurological impairment in childhood (Table 3). We present two males with a novel phenotype that includes prenatal-onset growth restriction, high MS-AFP and severe early neurological impairment with quadraparesis, Leber’s congenital amaurosis, hearing loss, central nervous system abnormalities and white matter changes. This previously unrecognized presentation expands the spectrum of PRPS1-related disorders and highlights the importance of PRPS1 in the development of the nervous system.
To understand the effect of the p.(Arg196Trp) mutation on the PRS-I enzyme function, we analyzed the molecular model for PRS-I. The crystal structure of PRS-I was determined by Li et al10 to consist of two subunits that form a homodimer. Three homodimers aggregate to form a hexamer. In this configuration, the catalytic active site and the allosteric regulatory site are formed by conserved residues from more than one subunit. The active site of PRS-I comprises the binding sites for ATP and ribose 5-phosphate. The side chain of the arginine at residue position 196 is pointed toward the bound ATP, and it is has been postulated that it is involved in the interaction with the pyrophosphate and in the stabilization of the transition state.10 The substitution of this conserved arginine with tryptophan is predicted to abolish this interaction and result in the decreased enzyme activity found in our patients.
Mutations in PRPS1 can have a different effect in proliferating cells and postmitotic cells, resulting in an array of phenotypic presentations.11 Brain MRI had been carried out in at least five patients with Arts syndrome in the past and showed no recognizable abnormalities.6 Autopsy of one individual who died at five and a half years of age revealed complete absence of myelinated axons in the posterior columns of the spinal cord, but no abnormalities were seen in the brain stem or in the gray and white matter of the cerebral and cerebellar hemispheres.2 Our patients presented with central nervous abnormalities including thin corpus callosum with the lack of isthmus and atrophied or hypoplastic optic chiasm. The white matter showed delayed maturation and thinning, signs that had not been previously recognized in this spectrum of disorders. In addition, our patients showed retinal dystrophy; as the retina is the tissue with the highest metabolic activity and that purines have an important role in phototransduction, it is not surprising that PRPS1 mutations lead to retinal degeneration.
We report for the first time an association between diabetes insipidus and PRPS1 deficiency. This is also likely related to severe GTP depletion in the brain. The paraventricular nucleus (PVN) and supraoptic nucleus (SON) of the hypothalamus are important mediators in homeostatic control including antidiuretic hormone secretion. G-protein-coupled receptors are expressed in the PVN and SON and have a major role eliciting rapid intracellular responses to regulate cell function.12
It has been demonstrated that purine nucleotide depletion can lead to the manifestations of the disorders caused by PRPS1 loss-of-function mutations. In lieu of neuronal tissue, erythrocytes provide a good model for brain cells.13 It was predicated but not demonstrated in the past that patients with Arts syndrome have purine nucleotide depletion.6, 14 Our patient had GTP and GDP depletion in erythrocytes, and the ATP+ADP/GTP+GDP ratio was increased. Depletion in GTP affects central neurological functions such as GTPases and dopamine synthesis.14 It is likely that GTP depletion is an important mechanism that results in the severe neurological manifestations in purine pathway disorders as demonstrated in the past for HGPRT deficiency (Lesch–Nyhan syndrome) and infantile PRS-1 superactivity.14, 15
The finding of orotic aciduria had not been previously described in patients with PRPS1 mutations. This elevation may be related to the fact that PRPP is necessary for the de novo biosynthesis of pyrimidine nucleotides, used to convert the nucleobase orotic acid into the nucleotide uridine monophosphate.13
Based on the analysis of the data from the exome sequence for these two patients, the reported novel mutation in PRPS1 appears to explain the entire phenotype. However, our patients show some biochemical differences with patients with the milder form of Arts syndrome that have been described previously. The levels of PRS-1 enzyme activity are extremely low in our patients but they are not undetectable as reported in other patients, and the levels of hypoxanthine are low but not undetectable. At present, there is no way to exclude completely the possibility that other mutation or mutations in coding or regulatory regions of the genome may be contributing to modify the phenotype. The recent description of a family with a PRPS1 mutation presenting in patients with overlapping symptoms of CMTX5 and Arts syndrome highlights the continuous disease spectrum of PRS-1 hypoactivity disorders.16 It has been suggested that the phenotypic presentation in males is determined by the exact PRPS1 mutation, its structural effect on the enzyme and the residual enzyme activity. The identification of additional patients with mutations affecting this residue may allow the development of better phenotype–genotype correlations.
Patients with Arts syndrome have shown encouraging response to a trial intervention with S-adenosylmethionine (SAM) as a substrate for the alternative purine synthesis pathway. SAM can replenish ATP and GTP independently of PRPP by direct conversion into adenine via the polyamine pathway. Additionally, methyltransferases convert SAM into S-adenosylhomocysteine, which can be transformed into adenosine and adenine by S-adenosylhomocysteine hydrolase.11
Recognition of this severe phenotype associated to PRS-1 deficiency expands the phenotype of syndromes caused by nucleotide depletion and provides important clues for its early prenatal consideration and diagnosis including IUGR and elevated levels of AFP. Several studies have demonstrated the association between a high MS-AFP and severe IUGR, neural tube and abdominal wall defects and fetal bowel obstruction.17, 18 AFP is a member of the albumin gene family and it is presumed to have a function in chemotaxis, oxygen free radical scavenging and lipid peroxidation.19 It is produced predominantly during fetal life when it reaches peak levels in the third trimester, declining rapidly after birth. Serum AFP is also elevated in certain neurodegenerative disorders such as ataxia telangiectasia, ataxia with oculomotor apraxia type 2 and some mitochondrial depletion syndromes. To date, it remains unclear if AFP is only a biochemical marker of the disease or if it contributes to the disease-causing mechanisms.19 This report adds severe PRS-1 deficiency to the group of disorders that need to be considered in the context of elevated MS-AFP.
The availability of a potential therapeutic intervention to ameliorate the symptoms by increasing the availability of purine nucleosides highlights the importance of achieving a timely molecular and biochemical diagnosis in infants with this presentation. Identification of cases with prenatal-onset manifestations may allow the initiation of therapeutic interventions early in the disease course.
References
Becker MA : Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog Nucleic Acid Res Mol Biol 2001; 69: 115–148.
Arts WF, Loonen MC, Sengers RC, Slooff JL : X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann Neurol 1993; 33: 535–539.
Kim HJ, Sohn KM, Shy ME et al: Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5). Am J Hum Genet 2007; 81: 552–558.
Lalwani AK, Brister JR, Fex J et al: A new nonsyndromic X-linked sensorineural hearing impairment linked to Xp21.2. Am J Hum Genet 1994; 55: 685–694.
Roessler BJ, Nosal JM, Smith PR et al: Human X-linked phosphoribosylpyrophosphate synthetase superactivity is associated with distinct point mutations in the PRPS1 gene. J Biol Chem 1993; 268: 26476–26481.
de Brouwer AP, Williams KL, Duley JA et al: Arts syndrome is caused by loss-of-function mutations in PRPS1. Am J Hum Genet 2007; 81: 507–518.
McVean GA : for The 1000 Genomes Consortium: An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Smith PK, Krohn RI, Hermanson GT et al: Measurement of protein using bicinchoninic acid. Anal Biochem 1985; 150: 76–85.
Bierau J, van Gennip AH, Helleman J, van Kuilenburg AB : The cytostatic- and differentiation-inducing effects of cyclopentenyl cytosine on neuroblastoma cell lines. Biochem Pharmacol 2001; 62: 1099–1105.
Li S, Lu Y, Peng B, Ding J : Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem J 2007; 401: 39–47.
de Brouwer AP, van Bokhoven H, Nabuurs SB, Arts WF, Christodoulou J, Duley J : PRPS1 mutations: four distinct syndromes and potential treatment. Am J Hum Genet 2010; 86: 506–518.
Hazell GG, Hindmarch CC, Pope GR et al: G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei – serpentine gateways to neuroendocrine homeostasis. Front Neuroendocrinol 2012; 33: 45–66.
Duley JA, Christodoulou J, de Brouwer AP : The PRPP synthetase spectrum: what does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids 2011; 30: 1129–1139.
Camici M, Micheli V, Ipata PL, Tozzi MG : Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem Int 2010; 56: 367–378.
Simmonds HA, Fairbanks LD, Morris GS, Webster DR, Harley EH : Altered erythrocyte nucleotide patterns are characteristic of inherited disorders of purine or pyrimidine metabolism. Clin Chim Acta 1988; 171: 197–210.
Synofzik M, Muller vom Hagen J, Haack T et al: X-linked Charcot–Marie–Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: evidence from a family with a novel PRPS1 mutation. Orphanet J Rare Dis 2014; 9: 24.
Bartha JL, Illanes S, Gonzalez-Bugatto F, Abdel-Fattah SA, Mizejewski GJ, Soothill PW : Maternal serum transformed alpha-fetoprotein levels in women with intrauterine growth retardation. Fetal Diagn Ther 2007; 22: 294–298.
Gagnon A, Wilson RD, Audibert F et al: Obstetrical complications associated with abnormal maternal serum markers analytes. J Obstet Gynaecol Can 2008; 30: 918–949.
Schieving JH, de Vries M, van Vugt JMG et al: Alpha-fetoprotein, a fascinating protein and biomarker in neurology. Eur J Paediatr Neurol 2013; 18: 243–248.
Acknowledgements
We thank the family for their ongoing participation and patience through this difficult diagnostic journey; FORGE Canada Consortium: Finding of Rare Disease Genes in Canada; Steering Committee: Kym Boycott (leader; University of Ottawa), Jan Friedman (co-lead; University of British Columbia), Jacques Michaud (co-lead; Université de Montréal), Francois Bernier (University of Calgary), Michael Brudno (University of Toronto), Bridget Fernandez (Memorial University), Bartha Knoppers (McGill University), Mark Samuels (Université de Montréal), Steve Scherer (University of Toronto); Janet Marcadier (Clinical Coordinator) and Chandree Beaulieu (Project Manager) for their contribution to the infrastructure of the FORGE Canada Consortium. We acknowledge the contribution of the high-throughput sequencing platform of The Centre for Applied Genomics, Toronto, Canada. This manuscript is dedicated to the memory of RC. This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research and the Ontario Genomics Institute (OGI-049). Additional funding was provided by Genome Quebec, Genome British Columbia and the McLaughlin Centre.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Al-Maawali, A., Dupuis, L., Blaser, S. et al. Prenatal growth restriction, retinal dystrophy, diabetes insipidus and white matter disease: expanding the spectrum of PRPS1-related disorders. Eur J Hum Genet 23, 310–316 (2015). https://doi.org/10.1038/ejhg.2014.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.112
This article is cited by
-
Direct stimulation of de novo nucleotide synthesis by O-GlcNAcylation
Nature Chemical Biology (2024)
-
A novel mutation in gene of PRPS1 in a young Chinese woman with X-linked gout: a case report and review of the literature
Clinical Rheumatology (2020)
-
Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases
Scientific Reports (2016)
-
Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes
Nature Reviews Endocrinology (2016)
-
Genetic Testing for Deaf and Hard of Hearing Individuals: Genetic Counseling
Current Genetic Medicine Reports (2016)

{kind=link}