
Abstract
The conventional approach to identifying the defective gene in a family with an inherited disease is to find the disease locus through family studies. However, the rapid development and decreasing cost of next generation sequencing facilitates a more direct approach. Here, we report the identification of a frameshift mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta (AI). Whole-exome sequencing of three affected family members and subsequent filtering of shared variants, without prior genetic linkage, sufficed to identify the pathogenic variant. Simultaneous analysis of multiple family members confirms segregation, enhancing the power to filter the genetic variation found and leading to rapid identification of the pathogenic variant. LAMB3 encodes a subunit of Laminin-5, one of a family of basement membrane proteins with essential functions in cell growth, movement and adhesion. Homozygous LAMB3 mutations cause junctional epidermolysis bullosa (JEB) and enamel defects are seen in JEB cases. However, to our knowledge, this is the first report of dominant AI due to a LAMB3 mutation in the absence of JEB.
Similar content being viewed by others

Introduction
Amelogenesis imperfecta (AI) is the collective term given to a genetically heterogeneous group of inherited defects of tooth enamel biomineralisation. Typically all teeth of the primary and secondary dentitions are affected, with marked variations in clinical phenotypes that are influenced by the underlying genotypes.1 The impact of AI on affected individuals and their families is considerable.2 To date, mutations in seven genes (AMELX, MIM 30039; ENAM, MIM 606585; KLK4, MIM 603767; MMP20, MIM 604629; FAM83H, MIM 611927; WDR72, MIM 613214 and C4orf26, MIM 614829) have been shown to cause either autosomal dominant, autosomal recessive or X-linked AI in the absence of other cosegregating abnormalities.3, 4, 5, 6, 7, 8, 9
Methods for identifying the defective gene in any given disorder generally involve identification of a disease locus followed by candidate gene sequencing. More recently next generation sequencing (NGS) has led to a hypothesis-null method of mutation identification through complete sequencing of exons at a locus in a single affected individual. With this approach a more manageable number of variants are highlighted after bioinformatic filtering than would be expected if examining the entire exome. These are then prioritised for segregation analysis, determination of frequency in control samples, analysis of evolutionary conservation and likely impact on protein function in order to determine the pathogenic variant.
Here, we report a further simplification of this approach, leading to the identification of a frameshift mutation in LAMININ-β3 (LAMB3; MIM 150310) in a family segregating non-syndromic dominant hypoplastic AI. Whole-exome NGS of three affected individuals, without prior genetic linkage, proved sufficient to identify the causative change.
Patients and methods
Members of the family AI-17, originating from Ireland, were recruited in accordance with the principles of the declaration of Helsinki, with informed consent and ethical approval. DNA samples were obtained from family members using either Oragene DNA sample collection kits (DNA Genotek, Kanata, ON, Canada) or via venous blood samples using conventional techniques. Screening of genes known to cause dominant AI (ENAM and FAM83H) was performed as previously described.10 Whole-exome sequencing was performed on a HiSeq2000 using the Nimblegen v2 chip by Otogenetics Corporation (Norcross, GA, USA). Resulting data were analysed and annotated using the DNAnexus (https://www.dnanexus.com/) data storage and analysis facility. Additional bioinformatic analysis of variants was performed using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/). The presence of potential variants was confirmed and segregation checked by PCR and Sanger sequencing using standard protocols. A multi-point Lod score was generated using Superlink Online (http://bioinfo.cs.technion.ac.il/superlink-online/).
Results
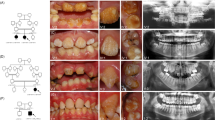
The proband of family AI-17 (arrowed in Figure 1a) presented with irregular hypoplastic AI and an autosomal dominant family history spanning four generations. Affected family members had experienced many symptomatic (eg, frequent pain) and aesthetic problems with their teeth (Figure 1b). Some individuals had received extensive restorative dental care and required multiple dental extractions. Review of the medical histories revealed no other health problems cosegregating with AI. In particular, no fragility, blistering or other skin problems were noted.

(a) Pedigree of family AI-17 illustrating segregation of the wild-type (+) and variant (−) alleles for the LAMB3 variant with the disease phenotype. The proband is marked with an arrow. Individuals who were whole-exome sequenced are marked with an asterix. (b) Clinical phenotype observed in the proband at 3 years of age. A generalised, irregular hypoplastic AI is observed with small islands of enamel evident on close inspection (arrows). These are absent from where the teeth occlude indicating that the clinical phenotype has probably been modified by post-eruptive changes. The panoramic radiograph of the proband confirms that the enamel is hypoplastic on formation. (c) Electropherogram showing the LAMB3 mutation and wild-type sequence. (d) Location of the LAMB3 change with respect to the Laminin-5 heterotrimer (based on Nakano et al).28
The known dominant AI genes ENAM and FAM83H were screened and excluded by PCR and Sanger sequencing in affected individual III:2. We then hypothesised that whole-exome sequencing of three distantly related, affected family members could allow the pathogenic mutation to be identified, without any prior mapping of a locus. Exonic DNA from individuals III:2, III:3 and IV:4 (indicated with * in Figure 1a) was therefore captured using a Nimblegen v2 exome reagent and clonally sequenced on a HiSeq2000 sequencer. The data generated are summarised in Supplementary Table 1.
The variants identified in each sample were filtered to exclude those >50 bp from an exon; known polymorphisms (variants listed in versions of dbSNP up to and including dbSNP129 or those with a minor allele frequency (MAF) >1%); and any variants, which did not pass standard Genome Analysis Toolkit variant filters. Total variants remaining after each of these stages of filtering are presented in Supplementary Table 2. Next, homozygous changes were excluded in light of the clear dominant inheritance in the family, and the remaining variants in each sample were compared to identify changes shared by all the three affected individuals sequenced. At this stage, any variant not within an exon or consensus-splice sequence was excluded. In addition, two of the remaining variants were retested using Sanger sequencing and found to be absent, and were therefore assumed to have been artefacts of NGS. This left twelve variants to take forward for further analyses (Table 1).
Of these, eight were present in dbSNP with a MAF of >0.1% and were therefore considered unlikely to be the cause of this rare dominant disease, given that the frequency of all forms of AI is estimated to be only ∼1/14 000 in the genetically heterogeneous US population.11 The MAF for a ninth variant was unknown but its presence in both the Watson and Venter genomes suggests it is a benign SNP present in the normal population. One further variant was predicted to be non-pathogenic by bioinformatic prediction software PolyPhen2. A 3 bp in-frame deletion in the HOMEZ gene (p.D547del; rs148005528) was excluded as it occurs within a run of eight aspartic acid residues, and four other 3 bp deletion SNPs occur within this run, each removing a single aspartic acid residue, suggesting that the deletion of a single residue is non-pathogenic.
This left only a single variant, a frameshift in the LAMB3 gene (NM_000228—c.3392_3393insG; p.E1133Gfs*27) on chromosome 1q32 (Figure 1c), as the potential cause of the disease phenotype. Homozygous LAMB3 mutations cause junctional epidermolysis bullosa (JEB) and enamel developmental defects are a universal finding in JEB, although severity and clinical expression vary widely.12 This variant was therefore considered a strong candidate for the pathogenic mutation in this family. Further investigation of the exome sequences of affected members of AI-17 confirmed that no variants were present in the collagen type XVII, alpha 1 gene (COL17A1), mutations in which can act in concert with LAMB3 mutations to cause digenic JEB.13 Sequencing of the LAMB3 variant using primers F:5′-GTTGGGAGTCTTGGGGAGTC-3′ and R:5′-AAGCATTCCAACCCAATCTG-3′ in all available family members revealed perfect segregation with the disease phenotype (Figure 1a). Furthermore, the variant was excluded from 180 Caucasian control individuals. A Lod score of 2.3 was achieved using the LAMB3 variant as a genetic marker, assuming 100% penetrance and an allele frequency of 0.0001.
Discussion
Current methods of gene identification generally require the mapping of a locus prior to Sanger sequencing or NGS. However, the wealth of data generated by NGS allow a range of techniques to be performed on just a single data set, leading to a shift in the methodological process of identifying disease-causing gene mutations. Here, we show that by subjecting the more distantly related affected individuals within a pedigree to whole-exome sequencing, the requirement for locus mapping is circumvented. Using this method, we were able to identify a novel heterozygous frameshift mutation in LAMB3, encoding a subunit of Laminin-5, as a cause of dominant hypoplastic AI.
Recessive mutations in LAMB3 are already known to cause JEB, characterised by intense skin blistering arising within the lamina lucida of the basement membrane, caused by a failure of hemidesmosome formation between the epidermis and lamina densa.14 Laminins are a family of network-forming trimeric proteins that are major constituents of the basal lamina, an extracellular matrix layer of the basement membrane. Laminin-5 consists of three polypeptides, alpha-3, beta-3, and gamma-2, encoded by the genes LAMA3 (MIM 600805), LAMB3, and LAMC2 (MIM 150292), respectively. Recessive mutations in these genes, and also in COL17A1 (MIM 113811), cause Herlitz (severe) and non-Herlitz (milder) JEB (MIM 226700),15 with LAMB3 mutations accounting for around 80% of cases.16 Digenic JEB has also been noted, in which compound heterozygous changes in COL17A1 combine with a heterozygous LAMB3 variant to cause JEB, though any two of the mutations described do not suffice to cause the disease.13
Most JEB cases also have enamel defects, including those due to recessive LAMB3 mutations, though these defects vary in severity.12 Furthermore, some heterozygous carriers of COL17A1 and LAMA3 mutations display an AI phenotype, though it is not common.17, 18, 19 No such cases have been reported for LAMB3 heterozygotes. Additionally, Col-17 and Lama3 knockout mice have defects in amelogenesis, with further examination revealing defects in Tomes’ processes causing disturbed enamel matrix secretion and malformed enamel rods.20, 21 While a Lamb3 mouse model exists, recessive mutants died within 24 h of birth and no examination of teeth was performed.22
The mutation identified in this study is a premature termination codon in the final exon of LAMB3. Nonsense mutations in the last exon of a gene are not thought to cause nonsense-mediated decay (NMD) as an exon/exon junction upstream of the mutation is required to bind NMD initiation factors.23 It therefore seems likely that a mutated protein will be expressed, with the C-terminal 40 amino acids of this 1172 amino-acid protein replaced by 27 incorrect amino acids. The C-terminus of LAMB3 primarily consists of an α-helical domain responsible for Laminin-5 subunit assembly24 (Figure 1d). The phenotype observed may therefore be a result of gain of function due to the mutant protein partially inhibiting the assembly of the heterotrimeric Laminin-5 complex. Interestingly, there is no evidence in the literature that parents of JEB children with LAMB3 mutations, themselves heterozygous carriers of LAMB3 mutations, have AI. However, many of the mutations described in JEB cases are nonsense mutations near the amino-terminus, which are more likely than the mutation described herein to cause a complete knockout of the gene. It therefore seems reasonable to speculate that the mutation described herein is unusual in its effect, acting as a dominant gain of function mutation to cause AI, while the majority of LAMB3 mutations are knock-outs, causing no phenotype in carriers while manifesting as JEB with enamel defects in homozygotes. Dental examinations in carrier parents of JEB cases due to a range of LAMB3 mutations would further test this hypothesis.
Amelogenesis involves three ameloblast stages; pre-secretory, secretory and maturation. In mice, Laminin-5 expression is observed mainly within secretory ameloblasts, particularly in Tomes’ processes, and in the enamel matrix, highlighting its requirement in the secretory stage of amelogenesis.25, 26 This correlates with the hypoplastic AI phenotype due to a reduced enamel matrix volume observed in the family investigated. Furthermore, Laminin-5 has been shown to be located on the apical and basal edge of secretory ameloblasts, suggesting it is required to anchor ameloblasts to the enamel matrix.25, 27 A defect in Laminin-5 is likely to lead to failure of transport across the cell-enamel matrix junction. Additionally, correct anchoring and arrangement of ameloblasts is required for crystal rod orientation, structure and mineralisation.26 Loss of Laminin-5 function is therefore likely to interrupt the secretory stage of amelogenesis, leading to a reduction in the volume of enamel matrix and impaired crystal rod growth.
In summary, we have identified a frameshift mutation at the extreme C-terminus of LAMB3, an integral member of the Laminin-5 heterotrimer, as a cause of dominant hypoplastic AI. The AI phenotype observed is consistent with a failure in the secretory stage of amelogenesis due to poor ameloblast adhesion to the secreted enamel matrix. In addition, we have shown that whole-exome sequencing of three distantly related family members can suffice to identify the causative mutation in a dominant family.
References
Bailleul-Forestier I, Molla M, Verloes A, Berdal A : The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. Eu J Med Genet 2008; 51: 273–291.
Coffield KD, Phillips C, Brady M, Roberts MW, Strauss RP, Wright JT : The psychosocial impact of developmental dental defects in people with hereditary amelogenesis imperfecta. J Am Dent Assoc 2005; 136: 620–630.
Lagerstrom M, Dahl N, Nakahori Y et al: A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 1991; 10: 971–975.
Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ : Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Human Mol Genet 2001; 10: 1673–1677.
Hart PS, Hart TC, Michalec MD et al: Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 2004; 41: 545–549.
Kim JW, Simmer JP, Hart TC et al: MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 2005; 42: 271–275.
Kim JW, Lee SK, Lee ZH et al: FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Human Genet 2008; 82: 489–494.
El-Sayed W, Parry DA, Shore RC et al: Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Human Genet 2009; 85: 699–705.
Parry DA, Brookes SJ, Logan CV et al: Mutations in c4orf26, encoding a Peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am J Human Genet 2012; 91: 565–571.
Wright JT, Torain M, Long K et al: Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells, Tissues, Organs 2011; 194: 279–283.
Witkop CJ, Sauk JJ Jr in: Oral Facial Genetics, eds Stewart R, Prescott G, 1976; 151–226 CV Mosby Company: St Louis.
Wright JT, Johnson LB, Fine JD : Development defects of enamel in humans with hereditary epidermolysis bullosa. Archiv Oral Biol 1993; 38: 945–955.
Floeth M, Bruckner-Tuderman L : Digenic junctional epidermolysis bullosa: mutations in COL17A1 and LAMB3 genes. American journal of human genetics 1999; 65: 1530–1537.
Schneider H, Muhle C, Pacho F : Biological function of laminin-5 and pathogenic impact of its deficiency. Eur J Cell Biol 2007; 86: 701–717.
Varki R, Sadowski S, Pfendner E, Uitto J : Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet 2006; 43: 641–652.
Pulkkinen L, Meneguzzi G, McGrath JA et al: Predominance of the recurrent mutation R635X in the LAMB3 gene in European patients with Herlitz junctional epidermolysis bullosa has implications for mutation detection strategy. J Investig Dermatol 1997; 109: 232–237.
McGrath JA, Gatalica B, Li K et al: Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol 1996; 148: 1787–1796.
Murrell DF, Pasmooij AM, Pas HH et al: Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J Investig Dermatol 2007; 127: 1772–1775.
Yuen WY, Pasmooij AM, Stellingsma C, Jonkman MF : Enamel defects in carriers of a novel LAMA3 mutation underlying Epidermolysis Bullosa. Acta dermato-venereologica 2012; 92: 695–696.
Asaka T, Akiyama M, Domon T et al: Type XVII collagen is a key player in tooth enamel formation. Am J Pathol 2009; 174: 91–100.
Ryan MC, Lee K, Miyashita Y, Carter WG : Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J Cell Biol 1999; 145: 1309–1323.
Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ : IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm Genome 1997; 8: 673–681.
Isken O, Maquat LE : The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev. Genetics 2008; 9: 699–712.
Aumailley M, El Khal A, Knoss N, Tunggal L : Laminin 5 processing and its integration into the ECM. Matrix Biol 2003; 22: 49–54.
Sahlberg C, Hormia M, Airenne T, Thesleff I : Laminin gamma2 expression is developmentally regulated during murine tooth morphogenesis and is intense in ameloblasts. J Dental Res 1998; 77: 1589–1596.
Yoshiba K, Yoshiba N, Aberdam D et al: Expression and localization of laminin-5 subunits during mouse tooth development. Dev Dyn 1998; 211: 164–176.
Aberdam D, Aguzzi A, Baudoin C, Galliano MF, Ortonne JP, Meneguzzi G : Developmental expression of nicein adhesion protein (laminin-5) subunits suggests multiple morphogenic roles. Cell Adhesion Commun 1994; 2: 115–129.
Nakano A, Chao SC, Pulkkinen L et al: Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Human genetics 2002; 110: 41–51.
Acknowledgements
We wish to thank the family used in this study. The project was funded by the Wellcome Trust Grant No. 093113. We are grateful to Dr David Parry for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Poulter, J., El-Sayed, W., Shore, R. et al. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet 22, 132–135 (2014). https://doi.org/10.1038/ejhg.2013.76
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.76
Keywords
This article is cited by
-
Autoimmune amelogenesis imperfecta in patients with APS-1 and coeliac disease
Nature (2023)
-
A novel nonsense variant in SLC24A4 causing a rare form of amelogenesis imperfecta in a Pakistani family
BMC Medical Genetics (2020)
-
Candidate gene sequencing reveals mutations causing hypoplastic amelogenesis imperfecta
Clinical Oral Investigations (2019)
-
Whole exome sequencing identifies an AMBN missense mutation causing severe autosomal-dominant amelogenesis imperfecta and dentin disorders
International Journal of Oral Science (2018)
-
Carriers with functional null mutations in LAMA3 have localized enamel abnormalities due to haploinsufficiency
European Journal of Human Genetics (2017)
