
Abstract
Congenital muscular dystrophies associated with brain malformations are a group of disorders frequently associated with aberrant glycosylation of α-dystroglycan. They include disease entities such a Walker–Warburg syndrome, muscle–eye–brain disease and various other clinical phenotypes. Different genes involved in glycosylation of α-dystroglycan are associated with these dystroglycanopathies. We describe a 5-year-old girl with psychomotor retardation, ataxia, spasticity, muscle weakness and increased serum creatine kinase levels. Immunhistochemistry of skeletal muscle revealed reduced glycosylated α-dystroglycan. Magnetic resonance imaging of the brain at 3.5 years of age showed increased T2 signal from supratentorial and infratentorial white matter, a hypoplastic pons and subcortical cerebellar cysts. By whole exome sequencing, the patient was identified to be compound heterozygous for a one-base duplication and a missense mutation in the gene B3GALNT2 (β-1,3-N-acetylgalactosaminyltransferase 2; B3GalNAc-T2). This patient showed a milder phenotype than previously described patients with mutations in the B3GALNT2 gene.
Similar content being viewed by others

Introduction
Aberrant glycosylation of α-dystroglycan is associated with a group of muscular dystrophies that range in severity from Walker–Warburg syndrome, a severe form of congenital muscular dystrophy with brain involvement to milder forms of limb girdle dystrophies.1, 2, 3, 4 Such disorders are caused by mutations in genes coding for glycosyltransferases or accessory proteins of glycosyltransferases including protein O-mannosyltransferase 1 (POMT1, MIM 607423), protein O-mannosyltransferase 2 (POMT2, MIM 607439), protein O-mannose β-1,2-N-acetylglucosaminyltransferase (POMGNT1, MIM 606822), fukutin (FKTN; MIM 607440), fukitin-related protein (FKRP, MIM 606596), acetylglucosaminyltransferase-like protein (LARGE, MIM 603590), isoprenoid synthase domain-containing protein (ISPD, MIM 614631), glycosyltransferase-like domain-containing protein 2 (GTDC2, MIM 147730), β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1, MIM 605517), GDP-mannose pyrophosphorylase B (GMPPB, MIM 615320), protein kinase-like protein sgk196 (SGK196, MIM 615247) and transmembrane protein 5 (TMEM5, MIM 605862). Additional genes link congenital muscular dystrophy with congenital disorders of glycosylation (DOLK, MIM 610746; DPM1, MIM 603503; DPM2, MIM 603564; and DPM3, MIM 605951). There is no clear correlation between the mutated gene and the clinical phenotype as all genes appear to act in the same pathway. It is rather the severity of a particular mutation than the affected gene that determines the severity of the clinical phenotype in dystroglycanopathies. All genes are known or predicted to alter the glycosylation of α-dystroglycan and therefore a reduction in α-dystroglycan glycosylation can be detected as a decrease in immunoreactivity with antibodies such as VIA4-1 and IIH6. However, many patients with dystroglycanopathy still remain genetically unidentified.5 Stevens et al6 recently reported that mutations in the gene β-1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2; MIM 610194) cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan with, in most cases, severe brain involvement.
By performing whole-exome sequencing in a patient with dystroglycanopathy, we identified compound heterozygous mutations in the B3GALNT2 gene. The clinical features and magnetic resonance imaging (MRI) findings were milder than in previously described patients with mutations in the B3GALNT2 gene.
Patients and methods
Patient
We describe a 5-year-old Swedish girl who was the first child to healthy and unrelated parents. After a normal pregnancy, she was born at term after an uneventful delivery with normal birth weight and an APGAR score of 6/10/10. A right-sided plexus brachialis palsy at birth that later normalized was noted. She had muscular hypotonia and muscle weakness since early age and her psychomotor development was generally delayed. She developed head control at 6 months of age, sat unsupported at 11 months of age and was able to walk unsupported at 3.5 years of age. She started to babble with two syllables at 11 months of age and said single words at 3.5 years of age. Psychological testing at 17 months of age with Griffith’s developmental scale I showed a global delay with a GQ score of 50 consistent with a moderate learning disability. At 6 years of age, she was able to speak single words and walked unsupported up to 3 km according to her parents. Growth in head circumference has been normal. Clinical examination showed mild muscle weakness with a negative Gower’s sign, gait ataxia and mild spasticity in the legs. In addition, she had increased tendon reflexes in the legs with bilateral ankle clonus and positive Babinski’s sign. Repeat cardiological investigations have been normal. Ophthalmological investigations at 4.5 years of age showed normal visual acuity, eye movements, refraction and fundus examination.
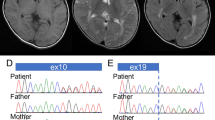
The level of serum creatine kinase has been consistently increased to a maximum of 647 IU/L (Reference value <180). MRI of the brain was performed at 3.5 years of age and demonstrated increased T2 signal from supratentorial and infratentorial white matter, a hypoplastic pons and subcortical cerebellar cysts (Figure 1). Supratentorial grey matter appeared normal without obvious cortical dysplasia and there were no signs of hydrocephalus.

MRI of the brain (1.5T) at 3.5 years of age. (a) Sagital T1 FLAIR (TR 2111.3 ms, TE 23.7 ms) images showed a thin and elongated pons. (b) Axial T2 (TR 2800 ms, TE 85 ms) and (c) coronal T2 FLAIR (TR 9002 ms, TE 161.4 ms) images showed hyperintense signalling, in periventricular white matter sparing the U-fibres, and in the infrantentorial white matter. (d) Axial T2 (TR 2800 ms, TE 85 ms) weighted images showed subcortical cerebellar cysts.
Muscle biopsy
Open muscle biopsy was performed of the quadriceps femoris, vastus lateralis muscle at 15 months of age. The specimen was frozen in propane chilled by liquid nitrogen and cryostat sections were subjected to routine staining techniques and immunohistochemical staining of dystrophin, sarcoglycans, merosin and α-dystroglycan (clone VIA4-1).
Genetic analysis
In order to identify the genetic cause in our patient, we performed whole-exome sequencing in the patient together with her parents (Figure 3a) with the SureSelect Human All Exon 50 Mb kit (Agilent Technologies, Santa Clara, CA, USA) and the HiSeq platform (Illumina, San Diego, CA, USA) as paired-end 100-bp reads. Alignment of reads, variant calling and filtering were performed as recently described.7 Sanger sequencing was performed using standard techniques of polymerase chain reaction amplicons with primers from genomic DNA to confirm the presence and identity of the variants in the B3GALNT2 gene after they had been identified by exome sequencing (primer sequences are available on request).
Results
Muscle biopsy
The muscle biopsy specimen showed no apparent morphological abnormalities (Figure 2a), whereas immunohistochemical analysis with antibodies against α-dystroglycan showed reduced labelling (Figure 2b). There was no increase in fibre size variability, no necrotic or regenerating muscle fibres, no increase of interstitial connective tissue, no increase in internally located nuclei and no cytoplasmic protein aggregates or other inclusion bodies. Immunostainings of dystrophin, sarcoglycans and merosin were all normal.

Quadriceps vastus lateralis muscle biopsy at the age of 15 months demonstrating normal morphology (a) and reduced immune-labelling of α-dystroglycan (b). (a) Hematoxilin and eosin, (b) immuno labelling of α-dystroglycan; inside is a normal control.
Genetic analysis
By bioinformatic analysis of the whole-exom data, we identified mutations in the B3GALNT2 (β-1,3-N-acetylgalactosaminyltransferase 2) gene (NM_152490.2). The patient was compound heterozygous for the variants g.chr1:235658059 (hg19), c.192dupT, p.(E65fs*) and g.chr1:235621957 (hg19), c.979G>A p.(D327N) (Figure 3b and c). Sanger sequencing was performed for verification of the mutations and the parents were identified to be carriers of one of the mutations each (Figure 3b and c). The substituted amino acid is conserved (UCSC genome browser) (Figure 3d). The variants have been submitted to the Leiden Muscular Dystrophy pages (http://www.LOVD.nl/B3GALNT2).

Pedigree and mutation analysis of B3GALNT2. (a) Pedigree of the family. (b) DNA sequencing chromatogram showing the c.192dupT mutation identified in exon 2, resulting in an premature stop-codon p.(E65fs*). (c) DNA sequencing chromatogram showing the c.979G>A mutation identified in exon 8, resulting in a missense mutation p.(D327N). Genomic positions for the mutations are according to hg19. (d) Protein sequence conservation of the missense mutation p.(D327N) (UCSC genome browser).
Discussion
In this report, we describe a case of congenital muscular dystrophy with brain involvement caused by novel B3GALNT2 mutations. Stevens et al6 speculate that after the addition of β-1,4-linked GlcNAc, B3GALNT2 completes the trisaccharide GalNAc-b1,3-GlcNAc-b1,4-Man. Mutations in this gene could reduce the amount of functional α-dystroglycan by preventing or impairing the formation of this glycan. The galactosyltransferase domain is located between amino acid position 307 and 457 in the protein. We speculate that the mutations identified in our patient affect the function of the galactosyltransferase domain indirectly and directly. One of the mutations was in exon 2, a duplication of a T affecting amino acid 65, resulting in a frame shift and a premature stop codon probably leading to nonsense-mediated mRNA decay. The second mutation was a missense mutation in exon 8, which changes the highly conserved amino acid 327 from an aspartic acid (Asp) to an asparagine (Asn) located inside the galactosyltransferase domain (Figure 3d).
In summary, whole-exome sequencing identified compound heterozygous mutations in the gene B3GALNT2. The clinical features with moderate central nervous system involvement, mild myopathy and no ophthalmological abnormalities were less severe than in previously reported patients with B3GALNT2 mutations. The MRI findings were also somewhat milder and we could not detect any obvious polymicrogyria or lissencephaly. The finding of normal histology in the muscle biopsy and reduced immunolabelling of α-dystroglycan demonstrates the importance of immunostaining in cases of undetermined muscle disorders, even in the absence of overt pathological changes. The findings in our patient broadens the spectrum of phenotypes associated with B3GALNT2 mutations and also indicates that B3GALNT2 mutations may be a frequent cause of α-dystroglycanopathy.
References
Godfrey C, Foley AR, Clement E, Muntoni F : Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011; 21: 278–285.
Clement E, Mercuri E, Godfrey C et al: Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 2008; 64: 573–582.
Hewitt JE : Abnormal glycosylation of dystroglycan in human genetic disease. Biochim Biophys Acta 2009; 1792: 853–861.
Cohn RD : Dystroglycan: important player in skeletal muscle and beyond. Neuromuscul Disord 2005; 15: 207–217.
Godfrey C, Clement E, Mein R et al: Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007; 130: 2725–2735.
Stevens E, Carss KJ, Cirak S et al: Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am J Hum Genet 2013; 92: 354–365.
Ohlsson M, Hedberg C, Bradvik B et al: Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 2012; 135: 1682–1694.
Acknowledgements
The study was supported by grants from the Swedish Research Council (project number 07122).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hedberg, C., Oldfors, A. & Darin, N. B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations. Eur J Hum Genet 22, 707–710 (2014). https://doi.org/10.1038/ejhg.2013.223
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.223
Keywords
This article is cited by
-
Elevated urinary excretion of free pyridinoline in Friesian horses suggests a breed-specific increase in collagen degradation
BMC Veterinary Research (2018)
-
Serial prenatal and postnatal MRI of dystroglycanopathy in a patient with familial B3GALNT2 mutation
Pediatric Radiology (2017)
-
B3GALNT2 mutations associated with non-syndromic autosomal recessive intellectual disability reveal a lack of genotype–phenotype associations in the muscular dystrophy-dystroglycanopathies
Genome Medicine (2017)
-
A nonsense mutation in B3GALNT2 is concordant with hydrocephalus in Friesian horses
BMC Genomics (2015)
-
Cerebellar Cysts in Children: a Pattern Recognition Approach
The Cerebellum (2015)
