
Abstract
Oligophrenin-1 (OPHN1) is one of at least seven genes located on chromosome X that take part in Rho GTPase-dependent signaling pathways involved in X-linked intellectual disability (XLID). Mutations in OPHN1 were primarily described as an exclusive cause of non-syndromic XLID, but the re-evaluation of the affected individuals using brain imaging displayed fronto-temporal atrophy and cerebellar hypoplasia as neuroanatomical marks. In this study, we describe clinical, genetic and neuroimaging data of a three generation Brazilian XLID family co-segregating a novel intragenic deletion in OPHN1. This deletion results in an in-frame loss of exon 7 at transcription level (c.781_891del; r.487_597del), which is predicted to abolish 37 amino acids from the highly conserved N-terminal BAR domain of OPHN1. cDNA expression analysis demonstrated that the mutant OPHN1 transcript is stable and no abnormal splicing was observed. Features shared by the affected males of this family include neonatal hypotonia, strabismus, prominent root of the nose, deep set eyes, hyperactivity and instability/intolerance to frustration. Cranial MRI scans showed large lateral ventricles, vermis hypoplasia and cystic dilatation of the cisterna magna in all affected males. Interestingly, hippocampal alterations that have not been reported in patients with loss-of-function OPHN1 mutations were found in three affected individuals, suggesting an important function for the BAR domain in the hippocampus. This is the first description of an in-frame deletion within the BAR domain of OPHN1 and could provide new insights into the role of this domain in relation to brain and cognitive development or function.
Similar content being viewed by others


Introduction
Intellectual disability (ID), formerly known as mental retardation (MR), is defined by a non-progressive reduction in cognitive abilities, which manifests before the age of 18 years and affects 2–3% of the general population.1 The etiopathological factors for ID are highly heterogeneous, including environmental aspects as well as established genetic causes, many of which represent X-chromosome-linked conditions (XLID). To date, more than 100 XLID genes were identified (http://www.ggc.org/research/molecular-studies/xlid.html), although mutation screens and linkage data strongly suggest that many more novel genes or mutational mechanisms remain elusive. Therefore, the significant percentage of non-syndromic XLID cases, in which no additional distinguishing features are present, turn the diagnostic approach into a great challenge with a high rate of underdiagnoses. As a consequence, endophenotyping, that is, the identification of subtle but characteristic clinical features, becomes key in the search for the underlying genetic cause in these patients.
Among the XLID-reported genes, at least seven encode proteins directly linked to Rho GTPase-dependent signaling pathways, as regulators (FGD1, ARHGEF6, OCRL1, GDI1, OPHN1) or effectors (FMR1, PAK3). Rho GTPases are a subfamily of small GTP-binding proteins that regulate spine morphogenesis and synapse development by functioning as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state. In their active conformation, Rho GTPases interact with specific effector molecules, which induce downstream signaling pathways that control a wide range of biological processes, including actin cytoskeletal reorganization, microtubule dynamics and membrane trafficking.2 These changes in neuronal morphology are crucial to the mechanisms of plasticity, learning and memory, so that inactivation of RhoGAP proteins might cause constitutive activation of their GTPase targets, which thus could result in XLID.
The oligophrenin-1 gene (OPHN1; MIM 300127), located at Xq12, was the first described Rho-linked ID gene, being identified after the molecular characterization of a X;12 balanced translocation in a female with mild ID.3, 4 Initially, mutations in this gene were reported to be responsible for non-syndromic XLID. Nevertheless, subsequent reports suggest that OPHN1 mutations result in a recognizable phenotype, which includes neuroradiological hallmarks such as cerebellar hypoplasia and ventriculomegaly, as well as subtle but characteristic facial features like strabismus and deep set eyes.5, 6
OPHN1 is expressed at low levels in all tissues, with a particularly higher expression in neurons during development and at later stages in highly plastic brain regions, such as the olfactory bulb and hippocampus.4, 7 OPHN1, localized both pre- and post-synaptically, is implicated in the regulation of dendritic spine morphology8, 9 and has a critical role in the activity-dependent maturation and plasticity of excitatory synapses by controlling their structural and functional stability.10 Indeed, Ophn1 deficiency in mouse displays similarities to the human phenotype and results in dendritic spine immaturity, ventricular enlargement and impaired spatial memory.9 Furthermore, knockout of Ophn1 in mice also reduces the endocytosis of synaptic vesicles and the post-synaptic AMPA receptor internalization, resulting in loss of long-term depression in the hippocampus.11
Herein, we describe clinical, genetic and neuroimaging findings from a three generation Brazilian family affected by XLID, resulting from a novel intragenic OPHN1 deletion (c.781_891del; r.487_597del), which is expected to lead to the excision of 37 amino acids (AA) from the highly conserved N-terminal BAR (Bin/amphiphysin/Rvs) domain. This in-frame deletion within the BAR domain could be responsible for the hippocampal alterations that were not detected in patients with a complete loss of OPHN1.
Patients and Methods
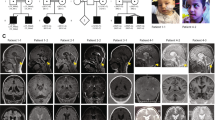
The propositus (III.2; Figure 1) was referred to the Human Genetics Service at the State University of Rio de Janeiro (Rio de Janeiro, Brazil) in 2009 because of an idiopathic familial history of ID and epilepsy, compatible with an X-linked inheritance pattern. The three generation family comprises 3 living affected males (II.3, III.2, III.4), 1 affected female (II.2) and 2 borderline individuals (one male (II.6) and one female (I.1)) in a total of 14 members available for testing (Figure 1).

OPHN1 deletion analysis in the family. (a) Family pedigree showing the segregation of the OPHN1 intragenic deletion ascertained through proband III.2. Solid squares represent boys with ID. Half solid square or circle indicates a borderline intellectual functioning, whereas the circle with a black dot represents an unaffected carrier female. The arrow points to the proband (III.2). ‘N’ indicates no deletion. ‘nt’ is ‘not available for testing’; (b) pictures of the affected males harboring the OPHN1 deletion; note some facial dysmorphies as ocular hypertelorism, deep set eyes, large ears and prominent chin; (c) pictures of the heterozygous females; note the same signs more or less evident.
For molecular analysis, genomic DNA was isolated from peripheral blood and cytogenetic evaluation was performed on cultured peripheral blood lymphocytes from the proband by standard methods. The Institutional Ethics Committee approved the research protocols and informed consent was obtained for all studied individuals.
FRAXA/FRAXE and multiplex ligation-dependent probe amplification (MLPA) analysis
Routine exclusion of trinucleotide repeat expansions in FMR1 and FMR2 genes was performed as previously described.12 The MLPA technique was applied for copy number variation analysis of 14 XLID genes (43 probes) on the X chromosome (Salsa kit P106-B1) according to the manufacturer’s recommendations (MRC Holland).
Array CGH and real-time quantitative PCR (qPCR) analysis
With the purpose of searching for submicroscopic imbalances along the entire X chromosome at a high resolution, we applied an oligo custom-designed X-chromosome-specific 244K array that covers the X chromosome exome, as well as its flanking 5′and 3′ untranslated regions (Agilent Technologies Inc., Santa Clara, CA, USA), as previously described.13 The slides were scanned on an Agilent DNA Microarray Scanner (Agilent Technologies Inc.) and images were extracted using the Feature Extraction software v9.1.3.1 (Agilent Technologies Inc.). The QC report was carefully examined to ensure proper hybridization and grid placement. The file generated by the Feature Extraction software was loaded into Agilent Genomics workbench Lite edition 6.0 software (Agilent Technologies Inc.) to allow data visualization. Z-score algorithm with a threshold of 6.0 was chosen to evaluate the distribution of data points and to identify copy number variations. All positions reported in this paper are based on the UCSC Genome Browser GRCh37/hg19 and NM_002547.2 was used for exon numbering.
Confirmation of the deletion was performed by regular PCR in males or real-time qPCR with the SYBR green chemistry on a 7500 Fast Real-time PCR system in females (Life Technologies, Foster City, CA, USA). Primers were designed using Primer 3 Plus software (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi) and RepeatMasker Documentation program (http://repeatmasker.genome.washington.edu). Sequences are available upon request. Reactions were performed in duplicate and a melting curve analysis was done to ensure specificity of each PCR product. Calculation of the relative gene copy number was achieved by the ΔΔCt method, using the PORCN locus at Xp11.23 as a normalizer. Results were confirmed in a second independent experiment. Fine mapping of the deletion was performed by iterative rounds of regular PCR.
Genomic DNA sequences of OPHN1 were loaded into the Vector NTI software (Life Technologies) to allow easy visualization of the position and extent of the aberration. PCR over the junction was performed with a combination of the forward primer annealing in the last normal region proximal to the deletion (5′-CGCAGTCAAA CACAAACCAG-3′) and the reverse primer annealing in the first normal region distal to the deletion (5′-TACTGGATCG GCACTTACAC C-3′). Bidirectional direct sequencing of the purified amplicon was performed with the BigDye Terminator kit on an ABI3130 automated sequencer (Life Technologies).
X-inactivation assay
For analysis of chromosome X inactivation (XCI) patterns among heterozygous females bearing the OPHN1 deletion, we proceeded on the androgen receptor (AR) methylation assay,14 using primers reported by Araujo et al15 for nested PCR. Allele profiles and areas under the curve for each allele were determined on an ABI310 Prism Genetic Analyzer (Life Technologies) and data were analyzed by GeneScan Analysis 3.7 and Genotyper 3.7 software (Life Technologies). Fluorescent peak areas representing true alleles were normalized for the occurrence of stutter products, and the XCI ratios were calculated as previously described.14
cDNA analysis
From all available individuals harboring the OPHN1 deletion (I.1, II.2, II.3, II.6, II.7 and III.2) as well as from control individuals, total RNA was extracted from whole blood stored in RNAlater solution (Life Technologies) with RiboPure blood kit (Ambion, Foster City, CA, USA). cDNA was generated starting from 170 to 900 ng of total RNA using the Superscript III First-Strand Synthesis System, containing random hexamer primers and Superscript III reverse transcriptase (Invitrogen). To investigate splice aberrations, we used a forward primer in exon 6 (5′-ACTGGATCGG CACTTACACC-3′) and a reverse primer in exon 8 (5′-GCTGTTGTTT GTATGGGAGG-3′) on 2 μl of cDNA on a Verity system (Life Technologies). PCR products were bidirectionaly sequenced using Big Dye Terminator on an ABI3130 automated sequencer (Life Technologies).
Neuroradiological data, EEG recording and cognitive assessment
All subjects presenting the OPHN1 deletion were imaged with a 1.5-T MR unit (HDXT, GE Healthcare, Milwaukee, WI, USA) with an eight-channel head coil. Routine images of the whole brain were obtained including sagittal FSE T1-weighted, axial T2 FLAIR (fluid-attenuated inversion recovery), axial diffusion weighted, coronal FSE T2-weighted, axial GRE T2-weighted and GRE 3D T1-weighted after contrast administration. Individuals I.1, II.2, II.3 and II.7 underwent routine scalp EEG under wakefulness and spontaneous superficial stages I and II non-REM sleep, whereas pediatric patients (III.2 and III.4) underwent induced sleep routine EEG. Individual II.6 refused to attend the EEG. Cognitive assessment was performed in individuals II.2 and II.3 using Raven matrices. The remaining affected individuals could not be tested because of the lack of comprehension (III.2) or refusal (I.1, II.6, III.4 and II.7).
Results
Clinical aspects
The propositus (III.2), a non-caucasian 8-year-old boy (year of birth 2004), was born by vaginal delivery in Rio de Janeiro/Brazil after a full-term uneventful pregnancy with a birth weight of 3000 g. Standard newborn screening was normal. Developmental milestones include head control at 3rd month, sitting position after 14th months and walking at the age of 30th months with a recognized underdeveloped speech. Epilepsy was noted since 6–8 months of age, whereas repetitive head movement was revealed at 14th months. Poor interaction with other children or adults was observed during his childhood, as well as psychomotor agitation and aggressiveness. Measure of length at 5.2 years demonstrated short stature (98 cm, below 3rd percentile). At the age of 8 years, the patient weighed 21 200 g (3/10 percentile) and his head circumference was 51 cm (next to 50 percentile). He used to walk just a little and then fall down, being attached to a wheelchair. He has no toilet training and asthma is also present. At the last physical exam (8 years old), the following features were noted (Figure 1): central prominence of forehead, bilateral, alternating and convergent strabismus, prominent root of the nose, deep set eyes, poor ocular contact, relative large ears (5.5 cm; 25/50 percentile), bluish macules in sclera, diastasis recti. Clinical investigation included normal results for behavioral audiometric studies, which assesses hearing acuity using unconditioned responses to sound (ie, reflexive and orienting behaviors). Tympanometry and acoustic reflex research indicated C curve at right side and A curve at left side (reflex present). A previous brain CT scan (4 years old) revealed ventricular and cisterna magna enlargement, suggesting cortical atrophy.
The proband’s youngest brother (III.4) has mild ID and his mother (II.2) is severely affected. The grandmother (I.1) has a borderline intellectual functioning, similar to that of one of her sons (II.6), who has a menial job at an open fair. His sister (II.7) is an apparently healthy female carrier. In addition, a second maternal uncle (II.3) of the proband exhibits severe ID. Clinical details of affected members of this family, except II.1 who died of unknown cause, can be found in Table 1. Features shared by affected male individuals include neonatal hypotonia, strabismus, prominent root of the nose, deep set eyes, hyperactivity and instability/intolerance to frustration. Both the proband (III.2) and his affected brother (III.4) experienced early onset seizures. III.2 presents both atonic and tonic-clonic seizures, whereas III.4 presents atonic seizures. Their mother (II.2) has also documented epilepsy (not infantile), presented as generalized tonic-clonic seizures.
Genetic analysis
A normal 550 band resolution karyotype was observed for the proband and expansions in FRAXA and FRAXE loci were ruled out. Because of the apparent X-linked inheritance pattern, we first performed MLPA to search for submicroscopic duplications/deletions in 14 XLID genes (PQBP1, TM4SF2, ARX, FMR1, GDI1, SLC6A8, RPS6KA3, ACSL4, DCX, IL1RAPL1, PAK3, ARHGEF6, AFF2 and OPHN1), which was negative. Next, we applied high-resolution X chromosome-specific oligo-array-CGH, which identified a subtle deletion of eight probes, encompassing exon 7 of the OPHN1 gene (ChrX:67 433 564–67 433 819; UCSC hg19; Figure 2a). This deletion was not detected by the commercial MLPA kit, as it only includes OPHN1 probes for exons 1, 3, 12 and 21.

Detection of the OPHN1 intragenic deletion. (a) X-chromosome oligo-array-CGH analysis plot. Cy3-labeled DNA of the proband was co-hybridized with Cy5-labeled DNA from a control male onto the array. The circle points to the deletion of eight subsequent probes. Note that the deletion is seen as an increased Cy5/Cy3 ratio. (b) RT-PCR analysis on RNA extracted from peripheral blood lymphocytes of several individuals, indicated below the gel. In the control sample (Co), the OPHN1 primer pair amplified a fragment of 251 bp from exon 6 to exon 8. In male patients, a band of 140 bp was obtained demonstrating the deletion of exon 7 at cDNA level. In the carrier females, two fragments were observed: one corresponding to the normal allele and the other referring to the deletion transcript. (c) Electropherogram obtained by sequencing the 140 bp PCR fragment showing the fusion of exon 6 to exon 8 due to the genomic OPHN1 deletion (c.781_891del; r.487_597del). The amino-acid sequence shown below the nucleotide sequence demonstrates that the removal of exon 7 yields an in-frame mutant transcript. (d) Ratios of abundance of the 140–251 bp bands between the three females harboring the deletion (I.1, II.2 and II.7). Dosage analysis was accomplished by the individual comparison of the band intensity of the deleted fragment with that of the normal one through GelQuant.Net software (http://biochemlabsolutions.com).
qPCR demonstrated that the deletion co-segregated with the ID phenotype in males (Figure 1a; II.3, II.6, III.2, III.4) and was absent in unaffected males (Figure 1a; II.4, III.1, III.3, III.6). In addition, the cognitively impaired mother (II.2) of the proband was shown to be a carrier of the deletion as was her mother (I.1) and her stepsister (II.7), who had normal intelligence. The three other tested healthy females (II.8, III.5, III.7) were negative for this aberration.
The absence of exon 7 on genomic level is predicted to result in an exon 7 lacking transcript. To test this assumption, we performed cDNA analysis from total RNA extracted from blood cells of affected individuals using OPHN1 primers in exon 6 and 8. Instead of the expected 251 bp PCR product, a band of 140 bp was obtained (Figure 2b). Indeed, sequence analysis revealed a transcript that misses exon 7 showing that exon 6 is spliced to exon 8 thereby removing 111 bp from the wild-type mRNA (Figure 2c, Supplementary Figure 1). This mutant transcript (c.781_891del; r.487_597del) was present in all affected males (II.3, II.6, III.2 and III.4).The carrier females (I.1, II.2 and II.7) also harbor this 140 bp fragment in addition to the wild-type 251 bp fragment. The ratio of abundance of the 140–251 bp band, although semi-quantitative, corresponds well with the clinical severity observed in these carrier females. Both bands show equal intensities for I.1 and II.2, which is associated with clinical characteristics. In II.7, the wild-type band (77%) is three times more intense compared with the 140 bp band (23%) reflecting the absence of clinical features in this carrier female (Figure 2d). Whereas the X-inactivation status in I.1 was not informative at the AR locus, those in the proband’s mother (II.2) and her stepsister (II.7) revealed random ratios of 71:29 and 26:74, respectively (Supplementary Table 1).
Clinical and genetic data from the proband were deposited in Decipher Consortium database (Patient 277638).
Bioinformatic analysis of the recombination
To precisely map the deletion breakpoints, we performed iterative rounds of PCR. When breakpoints regions were smaller than 2 kb at both sides of the deletion, we performed PCR over the junction revealing a product of about 800 bp in the proband but not in male controls. Sequencing of this band allowed us to define the junction of the deletion to the nucleotide level (ChrX:67 432 967–67 454 069; UCSC hg19). Bioinformatic analysis of the sequences flanking the deletion breakpoints with RepeatMasker demonstrated that the proximal breakpoint is located within an AluJB element in OPHN1 intron 6 and the distal one in a MIR3 element in intron 7. A 3-bp microhomology sequence (ACT) is present at the junction but without the introduction of deleted or added nucleotides, which points to a rearrangement driven by microhomology-mediated break-induced repair (MMBIR; Supplementary Figure 2).
Neuroimaging data, electrical patterns and cognitive assessment
MRI brain scans demonstrate large lateral ventricles, vermis hypoplasia and cystic dilatation of the cisterna magna in all affected males. In addition, hippocampal hypoplasia was present in individuals II.3 and III.2, whereas hippocampus verticalization was identified in individual III.4. Individual II.3 also exhibited microcephaly and mesencephalic verticalization (Figure 3). Among the carrier females, the intellectually normal aunt (individual II.7) did not present any neuroimaging alteration (data not shown), whereas the mother (individual II.2) exhibited periventricular cystic image, also seen in the proband, and hyperintensity lesions in the white matter, also noted in the grandmother (Figure 4). EEG recordings for individuals I.1, II.2, II.3 and II.7 showed normal background activity and physiologic elements of sleep were recorded. Patient II.7 showed one interictal discharge seen as a bilateral front-polar spike and wave. Moreover, hyperventilation caused a generalized slowing of her EEG that persisted until more than 20 s after its end. For children III.2 and III.4, induced sleep routine EEG recordings showed normal background activity corresponding to stage II non-REM sleep. III.4 recordings showed generalized spikes. Cognitive performance in the Raven test for both available individuals II.2 and II.3 was below the lower limit (percentile: 2; classification: V).

Neuroimaging scans of the males harboring the OPHN1 deletion. (a) Axial Flair weighted images show enlarged lateral ventricles (arrows) in patients II.3, III.2, III.4 and II.6. There is signal of hyperflow in the anterior horn of the left lateral ventricle of the patient III.4. (b) Sagital GRE 3D T1 images show vermis hypoplasia and cystic dilatation of the cisterna magna in patients II.3, III.2, III.4 and II.6. The patient II.3 also reveals microcephaly and a mesencephalic verticalization. (c) Coronal T2 weighted images show reduced volume of both hippocampus in patients II.3 and III.2 (hippocampus is shown by arrows). The left hippocampus in patient II.3 also shows a high signal intensity. Individual III.4 has verticalized hippocampus with normal volume.

Axial Flair weighted images from the carrier females. (a) Small cystic lesions (arrows) near the anterior horn of the left ventricles in the mother (II.2), which is also noted in the proband (III.2). (b) Focal white matter hyperintensities (arrows) near the atrium of right lateral ventricle and right semioval center in the mother (II.2) and left corona radiata in the grandmother (I.1). The aunt (II.7) did not present any neuroimaging alteration.
Discussion
In this study, we describe a novel intragenic deletion in OPHN1 (c.781_891del; r.487_597del) detected by X-array CGH that lead to an in-frame removal of 37 conserved amino acids in the BAR domain of OPHN1, which does not result in a loss of the protein. The highly conserved BAR domain (Supplementary Figure 3) is emerging as an important regulatory unit bridging membrane traffic and cytoskeletal dynamics. Over the past 15 years, a series of BAR domain-containing proteins linked to Rho GTPase signaling pathways have been characterized (for review see de Kreuk and Hordijk16).
OPHN1 is a Rho-GTPase-activating protein involved in XLID that comprises three main domains: a N-terminal Bin/Amphiphysin/Rvs (BAR) domain (19–225 AA) that binds curved membranes; a pleckstrin homology domain (265–370 AA) that is thought to confer membrane-binding specificity through interaction with phosphoinositides, and a central RhoGAP domain (380–572 AA) that regulates RhoA, Rac1 and Cdc42 and is able to stimulate the GTPase activity of small G protein. At its C-terminus, OPHN1 has also three proline-rich regions that act as putative SH3-binding sites for endocytic adaptor proteins.7, 17, 18 Functional analysis of OPHN1 in both neuronal and non-neuronal cells has demonstrated that the N-terminal segment including the BAR domain interacts directly with the GAP domain and inhibits its activity.7, 19 Recently, Elvers et al18 showed that the BAR domain guides OPHN1 to the plasma membrane, where it is able to interact with its substrate (active RhoGTPases), supporting the fact that changes in intracellular localization can contribute to GAP regulation. Furthermore, the authors also suggest that GAP domain may be regulated through the interaction with other proteins, such as 14–3–3 or filamin, which could account for BAR-mediated GAP inhibition. Nevertheless, it is not clear how the BAR domain binds to the GAP domain to inhibit its activity and how this inhibitory effect on GAP is abolished to allow OPHN1-GAP-mediated hydrolysis of Rho GTPases. In our patient, it is likely that the inhibitory effect of the mutant BAR domain on GAP is eliminated, permitting the hydrolysis. Another function attributed to the BAR domain is its role in the control of clathrin-mediated endocytosis.11
In the Database of Genomic Variants, the deletion reported in this study is not present indicating it is not a polymorphic variation. In relation to disease, there are six deletions involving OPHN1 described in Decipher. We disregarded two cases because of deletions >50 Mb encompassing many genes making genotype–phenotype correlation studies impossible. Among the four remaining cases, one represents a de novo 0.44 Mb deletion comprising the entire OPHN1 and YIPF6 genes in a male with cerebellar vermis hypoplasia, ID, seizures speech delay and strabismus (patient 2382). The other three patients (256 185, 256 487 and 258 853) harbor intragenic OPHN1 deletions ranging from 0.04 to 0.19 Mb. Two of them were identified in males (256 185 and 256 487) who inherited the loss from their apparently healthy mothers, but unfortunately no phenotypes were provided. The third was characterized in an ID female with a de novo OPHN1 deletion presenting early puberty and tall stature. The three intragenic OPHN1 deletions include multiple exons, which remove at least parts of the BAR domain. It is unknown, however, whether these deletions result in in-frame losses, as observed in our family.
The presence of microhomology at the junction of the deletion in our family could point to the rearrangement mechanism being non-homologous end joining or MMBIR. The DNA repair mechanism of non-homologous end joining, however, is prone to errors thereby generating an information scar at the junction, which is absent in our family. Therefore, we propose MMBIR here as substantial evidence has accumulated that the formation of microhomology junctions is often linked to DNA replication and repair, which is now seen as a major mechanism for change in copy number.20, 21 In this specific mechanism, replication fork stalling is repaired by strand invasion into non-homologous DNA based on microhomology followed by replication to the chromosome end.
According to the literature, 16 ID-related OPHN1 mutations were identified to date, including 2 translocations, 6 deletions, 3 nonsense, 3 frameshift and 2 splice site mutations.4, 5, 6, 22, 23, 24, 25, 26, 27, 28, 29 All but one of the reported mutations are thought to result in premature stop codons and the absence of any OPHN1 protein. As the exception, Pirozzi et al6 reported on a 2-bp OPHN1 deletion that abolishes a donor splicing site in intron 7 of OPHN1 in an Italian family segregating with ID and cerebellar hypoplasia. The deletion resulted in the inclusion of the initial 48 nucleotides of intron 7 in the mRNA, determining a mutant OPHN1 with 16 extra amino acids inserted in-frame in the N-terminal BAR domain.6 So, the OPHN1 intragenic deletion we present in this study is the first description of a deletion of conserved amino acids from the BAR domain, which could provide critical insight into the function of this domain.
The phenotype of affected members from different pedigrees presenting with loss-of-function OPHN1 mutations or an abnormal protein is not clinically distinguishable from each other and in comparison to the Brazilian family, mainly including mild to severe ID, vermis and/or hemispheric cerebellar hypoplasia, early seizures, strabismus and deep set eyes. However, we need to call particular attention to the hippocampal alterations presented by the proband (III.2), his younger brother (III.4) and his affected uncle (II.3; Figure 3), which are absent in the borderline affected uncle (II.6). It could explain the mild phenotype of this latter patient that preserve social skills. To our knowledge, hippocampal alterations have never been reported for mutations involving just the OPHN1 gene.
The proband (III.2), his younger brother (III.4) and their affected mother (II.2) have seizure episodes. Therefore, patients II.2 and III.2 had normal routine scalp EEGs, as may happen in up to 10% of patients with epilepsy, even when submitted to prolonged recordings and seems more common in extratemporal epilepsy patients such as ours.30 Patient III.4’s discharges are in accordance with generalized seizures.
Intrafamiliar phenotypic variability for OPHN1 mutations has previously been described for affected males5 as well as for carrier females, who usually present a mild phenotype with minor cognitive delay and subtle facial dysmorphies with or without brain abnormalities.3, 5, 23, 24, 25, 27 The absence of other discernible symptoms and signs among ID patients with OPHN1 mutations suggests that an OPHN1 deficiency may be compensated by functional redundancy with other Rho GTPase-related proteins in non-affected tissues. Alternatively, the structures of the brain where OPHN1 is expressed are characterized by high levels of plasticity and the brain may be more sensitive to loss of OPHN1 compared with other tissues.7
The three OPHN1 deletion carrier females in our report (individuals I.1, II.2 and II.7) present significant different phenotypes. The proband’s mother has severe ID, seizures and her dysmorphic facies resembles those seen in the affected males, whereas the proband’s grandmother has a borderline intelligence. Contrary, the aunt (II.7) is apparently cognitively unaffected. However, even she had never experienced seizures, EEG recording showed frontal epileptiform interictal activity (IEA). A number of brain diseases has been associated to IEA without epilepsy and IEA may sometimes precede overt seizures in weeks to years.31 Concerning the neuroimaging data, the profiles are also very different between the three carrier females. Although the brain hallmark of vermis hypoplasia is not observed, both mother and grandmother present hyperintensity lesions in the white matter. This phenotypic variability of carrier females most likely is due to different X inactivation patterns in the brain. Our data are in agreement with the five reported X inactivation studies in carrier females harboring loss-of-function mutations in OPHN1,5, 22, 24, 26, 28 which all found a random X inactivation pattern strongly suggesting that OPHN1 does not have a key role in early embryonic development, at least not in the hematopoietic lineage. Disease-associated CNVs on chromosome X among males are mostly inherited from their mothers, who usually do not present any clinical symptom and sign because of skewed X inactivation in favor of the normal chromosome X.28 Nonetheless, the random X inactivation in these studies was measured in blood and might not reflect the situation in the brain.
In conclusion, MRI testing of the vermis and/or hemispheric cerebellum should be considered for every patient with ID presenting with strabismus, seizures and deep set eyes. In parallel, a molecular screening for sequence mutations and structural genomic rearrangements of OPHN1 should be performed. Furthermore, careful comparison of the OPHN1 mutation with the observed phenotype can provide insight into the etiopathological mechanisms underlying XLID and the function of the affected protein domain.
References
Larson SA, Lakin KC, Anderson L, Kwak N, Lee JH, Anderson D : Prevalence of mental retardation and developmental disabilities: estimates from the 1994/1995 National Health Interview Survey Disability Supplements. Am J Ment Retard 2001; 106: 231–252.
Tolias KF, Duman JG, Um K : Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol 2011; 94: 133–148.
Bienvenu T, Der-Sarkissian H, Billuart P et al: Mapping of the X-breakpoint involved in a balanced X;12 translocation in a female with mild mental retardation. Eur J Hum Genet 1997; 5: 105–109.
Billuart P, Bienvenu T, Ronce N et al: Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 1998; 392: 923–926.
Al-Owain M, Kaya N, Al-Zaidan H et al: Novel intragenic deletion in OPHN1 in a family causing XLMR with cerebellar hypoplasia and distinctive facial appearance. Clin Genet 2011; 79: 363–370.
Pirozzi F, Di Raimo FR, Zanni G et al: Insertion of 16 amino acids in the BAR domain of the oligophrenin 1 protein causes mental retardation and cerebellar hypoplasia in an Italian family. Hum Mutat 2011; 32: E2294–E2307.
Fauchereau F, Herbrand U, Chafey P et al: The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol Cell Neurosci 2003; 23: 574–586.
Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L : The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci 2004; 7: 364–372.
Khelfaoui M, Denis C, van Galen E et al: Loss of X-linked mental retardation gene oligophrenin 1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci 2007; 27: 9439–9450.
Kasri NN, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L : The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev 2009; 23: 1289–1302.
Khelfaoui M, Pavlowsky A, Powell AD et al: Inhibition of RhoA pathway rescues the endocytosis defects in oligophrenin 1 mouse model of mental retardation. Hum Mol Genet 2009; 18: 2575–2583.
Santos CB, Pimentel MMG : The influence of expanded unmethylated alleles for FRAXA/FRAXE loci in the intellectual performance among Brazilian mentally impaired males. Int J Mol Med 2003; 12: 385–389.
Froyen G, Belet S, Martinez F et al: Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am J Hum Genet 2012; 91: 252–264.
Busque L, Paquette Y, Provost S et al: Skewing of X-inactivation ratios in blood cells of aging women is confirmed by independent methodologies. Blood 2009; 113: 3472–3474.
Araujo A, Ramos ES : Cryptic mosaicism involving a second chromosome X in patients with Turner syndrome. Braz J Med Biol Res 2008; 41: 368–372.
de Kreuk BJ, Hordijk PL : Control of Rho GTPase function by BAR-domains. Small GTPases 2012; 3: 45–52.
Nakano-Kobayashi A, Kasri NN, Newey SE, Van Aelst L : The Rho-linked mental retardation protein OPHN1 controls synaptic vesicle endocytosis via endophilin A1. Curr Biol 2009; 19: 1133–1139.
Elvers M, Beck S, Fotinos A, Ziegler M, Gawaz M : The GRAF family member oligophrenin1 is a RhoGAP with BAR domain and regulates Rho GTPases in platelets. Cardiovasc Res 2012; 94: 526–536.
Eberth A, Lundmark R, Gremer L et al: A BAR domain mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem J 2009; 417: 371–377.
Bauters M, Van Esch H, Friez MJ et al: Nonrecurrent MECP2 duplications mediated by genomic architecture driven DNA breaks and break-induced replication repair. Genome Res 2008; 18: 847–858.
Hastings PJ, Ira G, Lupski JR : A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 2009; 5: e1000327.
Tentler D, Gustavsson P, Leisti J et al: Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur J Hum Genet 1999; 7: 541–548.
Bergmann C, Zerres K, Senderek J et al: Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 2003; 126: 1537–1544.
Philip N, Chabrol B, Lossi AM et al: Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet 2003; 40: 441–446.
Chabrol B, Girard N, N'Guyen K et al: Delineation of the clinical phenotype associated with OPHN1 mutations based on the clinical and neuropsychological evaluation of three families. Am J Med Genet 2005; 138A: 314–317.
Zanni G, Saillour Y, Nagara M et al: Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology 2005; 65: 1364–1369.
Menten B, Buysse K, Vermeulen S et al: Report of a female patient with mental retardation and tall stature due to a chromosomal rearrangement disrupting the OPHN1 gene on Xq12. Eur J Med Genet 2007; 50: 446–454.
Froyen G, Van Esch H, Bauters M et al: Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum Mutat 2007; 28: 1034–1042.
Madrigal I, Rodríguez-Revenga L, Badenas C, Sánchez A, Milà M : Deletion of the OPHN1 gene detected by aCGH. J Intellect Disabil Res 2008; 52: 190–194.
Stüve O, Dodrill CB, Holmes MD, Miller JW : The absence of interictal spikes with documented seizures suggests extratemporal epilepsy. Epilepsia 2001; 42: 778–781.
Zivin L, Marsan CA : Incidence and prognostic significance of ‘epileptiform’ activity in the EEG of non-epileptic subjects. Brain 1968; 91: 751–778.
Acknowledgements
We thank the family members for their kind cooperation, ‘Centro Estadual de Diagnóstico por Imagem’ (SES, Rio de Janeiro, Brazil) for conducting the neuroimaging tests and Professor Paulo Luciano Gomes for helping in the EEG procedures. This work was supported by funds from CNPq (473824/2011-6), FAPERJ (E-26/103.215/2011), PPSUS-MS/CNPq/FAPERJ (E-26/110.765/2010) and CEPUERJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Santos-Rebouças, C., Belet, S., Guedes de Almeida, L. et al. A novel in-frame deletion affecting the BAR domain of OPHN1 in a family with intellectual disability and hippocampal alterations. Eur J Hum Genet 22, 644–651 (2014). https://doi.org/10.1038/ejhg.2013.216
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.216
