
Abstract
Autosomal-dominant idiopathic infantile nystagmus has been linked to 6p12 (OMIM 164100), 7p11.2 (OMIM 608345) and 13q31-q33 (OMIM 193003). PAX6 (11p13, OMIM 607108) mutations can also cause autosomal-dominant nystagmus, typically in association with aniridia or iris hypoplasia. We studied a large multigenerational white British family with autosomal-dominant nystagmus, normal irides and presenile cataracts. An SNP-based genome-wide analysis revealed a linkage to a 13.4-MB region on chromosome 11p13 with a maximum lod score of 2.93. A mutation analysis of the entire coding region and splice junctions of the PAX6 gene revealed a novel heterozygous missense mutation (c.227C>G) that segregated with the phenotype and is predicted to result in the amino-acid substitution of proline by arginine at codon 76 p.(P76R). The amino-acid variation p.(P76R) within the paired box domain is likely to destabilise the protein due to steric hindrance as a result of the introduction of a polar and larger amino acid. Eye movement recordings showed a significant intrafamilial variability of horizontal, vertical and torsional nystagmus. High-resolution in vivo imaging of the retina using optical coherence tomography (OCT) revealed features of foveal hypoplasia, including rudimentary foveal pit, incursion of inner retinal layers, short photoreceptor outer segments and optic nerve hypoplasia. Thus, this study presents a family that segregates a PAX6 mutation with nystagmus and foveal hypoplasia in the absence of iris abnormalities. Moreover, it is the first study showing detailed characteristics using eye movement recordings of autosomal-dominant nystagmus in a multigenerational family with a novel PAX6 mutation.
Similar content being viewed by others
Introduction
Infantile nystagmus is a genetically heterogeneous disorder consisting of involuntary oscillations of the eyes, often manifested within the first 6 months of life. Nystagmus has a prevalence of 1 in 1000.1 To date, two X-linked nystagmus loci (NYS1 (Xq26.2) and NYS5 (Xp11.4)) and three autosomal loci (NYS2 (6p12), NYS3 (7p11.2) and NYS4 (13q31-q33)),2, 3, 4, 5, 6, 7 have been mapped, and the NYS1 gene has been identified as FRMD7.2 Infantile nystagmus has also been described in association with afferent defects such as foveal hypoplasia, aniridia and iris hypoplasia.8, 9 Foveal hypoplasia is typically associated with PAX6 mutations,9 albinism10 and retinopathy of prematurity,11 whereas atypical forms of foveal hypoplasia have been associated with achromatopsia.12, 13, 14 Previous studies have also described foveal hypoplasia as being without an identifiable genetic cause.8, 15
We identified a large multigenerational white British family with a predominant phenotype of infantile nystagmus and presenile cataract segregating in an autosomal-dominant pattern. In this study, we have characterised the genotype and undertaken a linkage analysis and bidirectional Sanger sequencing. We report a novel PAX6 mutation and the phenotype associated with it.
Materials and methods
Phenotyping
The diagnosis of nystagmus was made based on clinical examination or eye movement recordings. Ophthalmic examination included slit lamp biomicroscopy, a dilated fundus examination, visual acuity testing and colour vision testing. Colour vision testing was done using the Farnsworth D-15 test. Electrophysiological testing was performed in subjects 634, 656, 668, 713 and 722, and consisted of a full-field electroretinogram response and visual-evoked potentials based on International Society for Clinical Electrophysiology of Vision standards.
Horizontal and vertical eye movement recordings were performed on six volunteers with nystagmus using an EyeLink I pupil tracker (250 Hz, SMI GmbH, Teltow, Germany). In five patients with a suspected torsional component to the nystagmus, a ‘Strabs VOG’ system was used to measure horizontal, vertical and torsional eye movements (50 Hz, SMI GmbH). Eye movements were recorded when the volunteers maintained fixation in primary positions and also in secondary positions at 20° (leftward, rightward, elevation and depression). Calibration of the eye movements was achieved offline.
We used an ultrahigh-resolution spectral domain OCT (SOCT Copernicus, OPTOPOL Technology S.A., Zawiercie, Poland) to obtain the tomograms in four affected family members. In order to capture the foveal region, a 7 × 7-mm scanning window was used, and this was centred at the fovea. The three-dimensional (3D) scan program (743 × 75; A scan × B scan) was used to obtain the region of interest. We have previously described the OCT acquisition and analysis protocol used for patients with nystagmus.16, 17
The study adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committees.
Genotyping
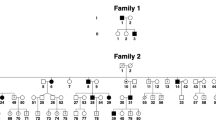
The pedigree was constructed and drawn using the Progeny Software, LLC (Delray Beach, FL, USA; www.progenygenetics.com) (Figure 1). Blood or saliva samples (Oragene Discover OG-250, DNA Genotek Inc., Ottawa, ON, Canada) were obtained with informed consent from 55 members (11 affected with nystagmus and 44 unaffected) of the family (Figure 1). DNA was extracted according to the manufacturer’s guidelines.

Pedigree of family with autosomal-dominant nystagmus. Additional clinical features included foveal hypoplasia and cataract. In addition to a clinical examination in some family members, we performed a high-resolution OCT and EMR.
Affymetrix GeneChip Human Mapping 10 K 2.0 SNP array (Affymetrix, Santa Clara, CA, USA) was used to generate genome-wide SNP data from individuals for a linkage analysis. Polymorphic markers—D11S902, D11S915, D11S904 and D11S935—were run to further define a region of linkage on the chromosome 11. Genetic marker and genotype data were stored, manipulated and error-checked using the genetic data management system (Progeny Software LLC, Delray Beach, FL, http://www.progenygenetics.com) before being exported to Allegro (version 2) for a fast multipoint linkage analysis.18 We assumed a dominant mode of inheritance with full penetrance and a disease gene frequency of 0.0001.
Primers were designed to amplify the coding exons and the intron–exon boundaries of the PAX6 gene (Accession ID: NG_008679.1) (Table 1). All the coding exons and splice junctions were Sanger-sequenced bidirectionally in two affected subjects (631 and 633). Sequence data were visualised and compared using the software ‘Sequencher’ (Gene Codes Corporation, Ann Arbor, MI, USA). The PAX6 transcript (NM_000280.4) was imported for variant calling and subsequent annotation as per the Human Genome Variation Society guidelines. Subsequent segregation analysis was performed using denaturing high-performance liquid chromatography. DNA from each enrolled family member was first amplified using the PAX6_3 primer set, and the resulting amplicon was digested with the SURVEYOR nuclease, a mismatch-specific nuclease (Transgenomic, Inc., Omaha, NE, USA). Mismatch in DNA fragments is formed when a heterozygous variant is present within the amplicon after multiple denaturation steps before digestion. Hence, control amplicons will remain undigested, whereas mutated amplicons will be digested into two fragments. The final digested products were run on denaturing high-performance liquid chromatography for fragment size discrimination. Variant data were submitted to the Leiden Open Variation Database.
Results
Phenotyping
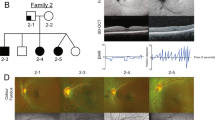
The clinical features associated with this family are outlined in Table 2. There was an autosomal-dominant inheritance of nystagmus, with X-linked inheritance ruled out by male-to-male transmission. Presenile cataracts co-segregated with nystagmus, except in the two youngest patients (Table 2). Cataracts occurred in the second to third decade. All examined patients had a normal iris structure (Figure 2a), with no features of aniridia or iris hypoplasia. Subjects 656 and 668 had blepharoptosis. A fundus examination revealed foveal hypoplasia, which was confirmed on OCT. The results of electroretinogram and visual-evoked potentials were normal for the tested subjects.

Examples of phenotypical characteristics of the anterior (A) and posterior segments (B). (A) All subjects had a normal iris structure. Cataract was present in some individuals, but not all affected family members with nystagmus had cataract. (B) The fundus photos and OCT displayed are from subject 668 (on the left) and 657 (on the right). Both fundus photos and OCTs show features of foveal hypoplasia. In subject 668, there is no foveal pit, whereas in subject 657, there is a rudimentary pit (a). In both individuals, there is an incursion of inner retinal layers posterior to the foveola (b), which is a hallmark of foveal hypoplasia. Both subjects also had shorter outer segments (c). We also noticed a thinner retinal nerve fibre layer (d) in the patients with PAX6 mutation.
The foveal OCT tomograms showed a number of abnormalities consistent with foveal hypoplasia and optic nerve hypoplasia, as follows: (a) the foveal pit was shallower or absent; (b) there was a continuation of inner retinal layers posterior to the foveola; (c) the outer segments at the fovea were shorter and (d) the retinal nerve fibre layer appeared thinner. There was some variability in foveal morphology between family members; for instance, subject 657 had a rudimentary foveal pit, whereas her mother (668) had fovea plana (Figure 2b).
Eye movement recordings (EMR) were obtained from six affected subjects (see Figure 1). All the subjects had conjugate nystagmus, although further analysis of the eye movement recordings showed large intrafamilial variability in the nystagmus characteristics (Table 3). The nystagmus oscillations were in different planes (horizontal, vertical and torsional). Subject 656 had a significant vertical component visible on clinical examination and also in eye movement recordings. Although subject 634 appeared to have only horizontal nystagmus on clinical examination, in eye movement recordings, it was evident that a small vertical component was also present. Interestingly, subject 657 had pendular nystagmus in a primary position, which changed to vertical jerk nystagmus in an eccentric gaze position. Examples of the nystagmus waveforms are shown in Figure 3.

Examples of the nystagmus waveform from different family members. Both horizontal (Hor) and vertical (Ver) traces are shown. Large intrafamilial variability of the nystagmus characteristics was seen.
Genotyping
We undertook a combination of SNP-based genome-wide analysis (Affymetrix mapping 10K SNP array) and microsatellite-based genotyping in 12 members. A maximum lod score of 2.93 was obtained with markers within a 13.4-MB region on chromosome 11p13 flanked by rs722490 and rs113634546. The PAX6 gene is located within this genomic interval, and hence was prioritised for mutation analysis.
Sequencing of the PAX6-coding exons (exons 4–13) and the flanking intron–exon boundaries in two affected family members detected a novel heterozygous c.227C>G variant in exon 8 (Figure 4a). This missense variant is predicted to substitute a postively charged arginine for the highly conserved proline at amino-acid position 76 (Figure 4b). We used Swiss-PdbViewer (DeepView) to model the effect19 of the amino-acid variation p.(P76R) on the 3D structure of the protein. The mutation alters a residue located within the paired box domain. This domain contains N-terminal and C-terminal subdomains termed PAI (amino-acid position 3–64) and RED (amino-acid position 79–131), respectively. The PAI and RED subdomains are connected via a linker subdomain (amino-acid position 64–79), which binds to DNA through the minor groove.20, 21 The substitution of the proline residue by a larger and more polar arginine residue at position 76 most likely destabilises the linker subdomain, affecting the normal functioning of the PAX6 protein (Figures 4c and d).

The heterozygous mutation detected in the family is shown in (a). The mutation results in amino-acid substitution p.(P76R). Multiple sequence alignment shows that the amino-acid position 76 is identical between different species and is located within invariant blocks of identical residues (b). The non-polar residues have a yellow background, whereas the blue, green and red background represents amino acids with a polar side chain. The 3D structure of the paired box domain adjacent to the DNA is shown in c, the proline residue has been highlighted. The mutation results in a disruption of the paired box domain and is likely to destabilise the protein due to steric hindrance as a result of the introduction of a polar and larger amino acid (arginine) (c and d).
Discussion
We have identified a causative PAX6 mutation in a large family with autosomal-dominant nystagmus and normal irides, providing evidence of a phenotypic heterogeneity associated with nystagmus in patients with PAX6 mutations. We have previously reported a phenotypic heterogeneity associated with nystagmus in families with FRMD7 mutations and also in patients with periodic alternating nystagmus.7, 22 These data suggest that the nystagmus phenotype is not defined solely by genotype.
Different neural substrates control horizontal and vertical gaze, and their selective disruptions could, in theory, explain nystagmus directionality. Most infantile forms of nystagmus, such as idiopathic nystagmus with or without a mutation in FRMD7 or nystagmus associated with albinism, are in the horizontal direction. Consistently with this, we have observed the expression of FRMD7 within the developing horizontal neural integrator (nucleus prepositus and medial vestibular nuclei).7, 22 In this family, we report horizontal, vertical and torsional nystagmus associated with a PAX6 mutation. Neuroimaging in patients with PAX6 mutations have revealed abnormalities in the cerebellar grey matter.23 Lesions on the cerebellum, in particular the flocculus, have been attributed to downbeat nystagmus,24, 25 and homozygous Pax6 knockout embryos have disrupted cellebellar cell migration and neurite extension.26 Thus, it is possible that multiple gaze control centres are affected in patients with PAX6 mutations, resulting in the nystagmus on various planes as compared with other patients with infantile nystagmus, who invariably have horizontal nystagmus.27 Furthermore, the postnatal development of the visual system may be affected by afferent abnormalities such as foveal hypoplasia, optic nerve hypoplasia and cataract, and contribute to the nystagmus phenotype.
We observed differences in the foveal structure between family members. The phenotypical heterogeneity of foveal morphology based on OCT is an interesting observation that has not been previously reported among affected members of the same family with PAX6 mutations. This suggests that foveal development is also not entirely predicted by genotype. Previous histological data suggest that the fovea continues to develop up to the age of 15–45 months.28 Thus, variations in postnatal development could account for this phenotypic heterogeneity. PAX6 is expressed within the developing human retina, in particular, the ganglion cell layer.29 We observed that the patients had a thinner retinal nerve fibre layer, consistent with optic nerve hypoplasia. This has previously been observed with PAX6 mutations.9, 30
The phenotype described in this study has some similarities to those of two families previously reported in the literature. Hanson et al30 described a family with autosomal-dominant nystagmus, congenital cataract, corneal epithelial changes and foveal hypoplasia (OMIM 136520). O'Donnell and Pappas31 reported a family with similar phenotypical characteristics, which included congenital nystagmus, presenile cataract, foveal hypoplasia and peripheral corneal pannus, which was dominantly inherited as part of this syndrome (OMIM 136520). The family reported by Hanson et al30 had a PAX6 mutation (p.G64V). However, genetic studies were not performed on the family reported by O'Donnell and Pappas31. The main difference in phenotype between our family and the aforementioned families is the normal corneal structure associated with the p.(P76R) variant.
Hingorani et al9 have shown in a large cohort of patients (n=43) that foveal hypoplasia is always associated with loss of function and C-terminal extension PAX6 mutations and seldom associated with missense mutations. Our results, combined with previous studies, however, support the association of foveal hypoplasia with missense mutations specifically altering amino-acid residues close to codon 76. In addition to this study, p.G72S, p.G73D and p.S74G were reported to be associated with foveal hypoplasia.9, 32, 33 The p.G73D variant was also associated with aniridia,33 whereas patients with the p.G72S variant had an abnormal iris structure.9 Gronskov et al34 reported iris hypoplasia, eccentric pupil, nystagmus and early cataract to be associated with the p.A79E variant; however, an additional variant was also identified—p.R208Q. The p.R208Q was inherited from the unaffected mother, hence its role in the resulting phenotype is unclear. PAX6 consists of two DNA-binding domains, the paired box domain and the homeodomain. The variants described above are all located within the paired box domain and, specifically, within the linker subdomain, and are thus likely to result in reduced DNA binding and altered transcriptional activation function as previously demostrated with the help of functional assays using paired box domain missense mutant proteins.30 Although modifier genes could also contribute to the phenotype, there have been consistent reports that missense mutations within the PAX6 domain linker region are associated with milder phenotypes. If modifiers are involved, it is plausible that they reside in the region tightly linked to or even within the paired box domain, as the phenotypes described in this study and by Dansault et al.32 are consistently observed in a number of individuals. Interestingly, the patient with the p.S74G variant had intellectual disabilities and epilepsy, and MRI revealed hypoplasia of the anterior commisure and an absent pineal gland. None of the subjects in this study had a history of epilepsy or intellectual disability; however, we have not performed MRI studies in our subjects to assess the brain structure.
Hingorani et al9 also reported that each of the 43 patients in her cohort had some form of iris anomaly (ranging from abnormal iris structure to no iris). Thus, although in retrospect, there were some phenotypic characteristics in our family consistent with a PAX6 mutation, the normal iris and corneal structure in all the affected individuals prompted us to perform a whole-genome SNP analysis before the candidate gene selection and sequencing. This study shows that in patients with autosomal-dominant nystagmus with variable characteristics, a screening of PAX6 for mutations should be considered even in the setting of a normal iris structure.
References
Stayte M, Reeves B, Wortham C : Ocular and vision defects in preschool children. Br J Ophthalmol 1993; 77: 228–232.
Tarpey P, Thomas S, Sarvananthan N et al: Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet 2006; 38: 1242–1244.
Cabot A, Rozet JM, Gerber S et al: A gene for X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome Xp11.4-p11.3. Am J Hum Genet 1999; 64: 1141–1146.
Klein C, Vieregge P, Heide W et al: Exclusion of chromosome regions 6p12 and 15q11, but not chromosome region 7p11, in a German family with autosomal dominant congenital nystagmus. Genomics 1998; 54: 176–177.
Ragge NK, Hartley C, Dearlove AM, Walker J, Russell-Eggitt I, Harris CM : Familial vestibulocerebellar disorder maps to chromosome 13q31-q33: a new nystagmus locus. J Med Genet 2003; 40: 37–41.
Kerrison JB, Arnould VJ, Barmada MM, Koenekoop RK, Schmeckpeper BJ, Maumenee IH : A gene for autosomal dominant congenital nystagmus localizes to 6p12. Genomics 1996; 33: 523–526.
Thomas MG, Crosier M, Lindsay S et al: The clinical and molecular genetic features of idiopathic infantile periodic alternating nystagmus. Brain 2011; 134: 892–902.
Thomas MG, Kumar A, Mohammad S et al: Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology 2011; 118: 1653–1660.
Hingorani M, Williamson KA, Moore AT, van Heyningen V : Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci 2009; 50: 2581–2590.
Oetting WS, King RA : Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat 1999; 13: 99–115.
Hammer DX, Iftimia NV, Ferguson RD et al: Foveal fine structure in retinopathy of prematurity: an adaptive optics Fourier domain optical coherence tomography study. Invest Ophthalmol Vis Sci 2008; 49: 2061–2070.
Thiadens AA, Somervuo V, van den Born LI et al: Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci 2010; 51: 5952–5957.
Thomas MG, Kumar A, Kohl S, Proudlock FA, Gottlob I : High-resolution in vivo imaging in achromatopsia. Ophthalmology 2011; 118: 882–887.
Thomas MG, McLean RJ, Kohl S, Sheth V, Gottlob I : Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br J Ophthalmol 2012; 96: 1232–1236.
Querques G, Bux AV, Iaculli C, Delle Noci N : Isolated foveal hypoplasia. Retina 2008; 28: 1552–1553.
Thomas MG, Kumar A, Thompson JR, Proudlock FA, Straatman K, Gottlob I : Is high-resolution spectral domain optical coherence tomography reliable in nystagmus? Br J Ophthalmol 2013; 97: 534–536.
Thomas MG, Kumar A, Kohl S, Proudlock FA, Gottlob I : High resolution in-vivo imaging in achromaptopsia. Ophthalmology 2011; 118: 882–887.
Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A : Allegro version 2. Nat Genet 2005; 37: 1015–1016.
Guex N, Peitsch MC : SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18: 2714–2723.
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO : Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev 1999; 13: 1263–1275.
Epstein J, Cai J, Glaser T, Jepeal L, Maas R : Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J Biol Chem 1994; 269: 8355–8361.
Thomas S, Proudlock FA, Sarvananthan N et al: Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain 2008; 131: 1259–1267.
Free SL, Mitchell TN, Williamson KA et al: Quantitative MR image analysis in subjects with defects in the PAX6 gene. Neuroimage 2003; 20: 2281–2290.
Ito M, Nisimaru N, Yamamoto M : Specific patterns of neuronal connexions involved in the control of the rabbit's vestibulo-ocular reflexes by the cerebellar flocculus. J Physiol 1977; 265: 833–854.
Baloh RW, Spooner JW : Downbeat nystagmus: a type of central vestibular nystagmus. Neurology 1981; 31: 304–310.
Engelkamp D, Rashbass P, Seawright A, van Heyningen V : Role of Pax6 in development of the cerebellar system. Development 1999; 126: 3585–3596.
Kumar A, Gottlob I, Mclean RJ, Thomas S, Thomas MG, Proudlock FA : Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus. Invest Ophthalmol Visual Sci 2011; 52: 2306–2313.
Hendrickson AE, Yuodelis C : The morphological development of the human fovea. Ophthalmology 1984; 91: 603–612.
Nishina S, Kohsaka S, Yamaguchi Y et al: PAX6 expression in the developing human eye. Br J Ophthalmol 1999; 83: 723–727.
Hanson I, Churchill A, Love J et al: Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet 1999; 8: 165–172.
O’Donnell FE Jr, Pappas HR : Autosomal dominant foveal hypoplasia and presenile cataracts. A new syndrome. Arch Ophthalmol 1982; 100: 279–281.
Dansault A, David G, Schwartz C et al: Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol Vis 2007; 13: 511–523.
Chao LY, Mishra R, Strong LC, Saunders GF : Missense mutations in the DNA-binding region and termination codon in PAX6. Hum Mutat 2003; 21: 138–145.
Gronskov K, Rosenberg T, Sand A, Brondum-Nielsen K : Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet 1999; 7: 274–286.
Acknowledgements
Ulverscroft Foundation, National Eye Research Centre and Howard Hughes Medical Institute. ECE is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Thomas, S., Thomas, M., Andrews, C. et al. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet 22, 344–349 (2014). https://doi.org/10.1038/ejhg.2013.162
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.162
Keywords
This article is cited by
-
A new association of PAX6 variation with Juvenile onset open angle glaucoma
Journal of Human Genetics (2023)
-
Variants in PAX6, PITX3 and HSF4 causing autosomal dominant congenital cataracts
Eye (2022)
-
Diagnostik, Klinik und Genetik kongenitaler Hornhauttrübungen
Der Ophthalmologe (2022)
-
PAX6 missense variants in two families with isolated foveal hypoplasia and nystagmus: evidence of paternal postzygotic mosaicism
European Journal of Human Genetics (2021)
-
Homozygous single nucleotide duplication of SLC38A8 in autosomal recessive foveal hypoplasia: The first Japanese case report
Documenta Ophthalmologica (2021)
