
Abstract
There is strong evidence that rare copy number variants (CNVs) have a role in susceptibility to autism spectrum disorders (ASDs). Much research has focused on how CNVs mediate a phenotypic effect by altering gene expression levels. We investigated an alternative mechanism whereby CNVs combine the 5′ and 3′ ends of two genes, creating a ‘fusion gene’. Any resulting mRNA with an open reading frame could potentially alter the phenotype via a gain-of-function mechanism. We examined 2382 and 3096 rare CNVs from 996 individuals with ASD and 1287 controls, respectively, for potential to generate fusion transcripts. There was no increased burden in individuals with ASD; 122/996 cases harbored at least one rare CNV of this type, compared with 179/1287 controls (P=0.89). There was also no difference in the overall frequency distribution between cases and controls. We examined specific examples of such CNVs nominated by case–control analysis and a candidate approach. Accordingly, a duplication involving REEP1-POLR1A (found in 3/996 cases and 0/1287 controls) and a single occurrence CNV involving KIAA0319-TDP2 were tested. However, no fusion transcripts were detected by RT-PCR. Analysis of additional samples based on cell line availability resulted in validation of a MAPKAPK5-ACAD10 fusion transcript in two probands. However, this variant was present in controls at a similar rate and is unlikely to influence ASD susceptibility. In summary, although we find no evidence that fusion-gene generating CNVs lead to ASD susceptibility, discovery of a MAPKAPK5-ACAD10 transcript with an estimated frequency of ∼1/200 suggests that gain-of-function mechanisms should be considered in future CNVs studies.
Similar content being viewed by others

Introduction
Copy number variants (CNVs) are deletions or duplications of chromosomal segments ranging from a few thousand to several million base pairs. A number of population-based studies have shown that this type of genomic variant can affect as much as 12% of the human genome.1 Many studies have gone on to demonstrate the importance of CNVs in determining human phenotypic variation and disease susceptibility.2, 3
The Autism Genome Project consortium (AGP) recently detected an excess of genes disrupted by rare CNVs in 996 individuals with autism spectrum disorder (ASD) in comparison to 1287 control subjects.4 This increased burden demonstrates the relevance of CNVs in susceptibility to this early-onset neurodevelopmental condition. Unlike some other studies, where exceptionally rare homozygous CNVs were seen in long tracts of homozygosity-by-descent,5 the AGP cohort contains very few consanguineous ASD pedigrees and therefore the vast majority of these rare CNVs are heterozygous.
Studies on both humans and mice have shown that CNVs can influence gene expression.6, 7 This has led many scientists to hypothesize that these heterozygous CNVs may influence susceptibility to neurodevelopmental disorders through a gene-dosage mechanism. For instance, it is easy to imagine how mild perturbations in axon guidance molecules could affect critical stages of brain development. Others have looked for evidence of an alternative mechanism whereby deletions might unmask recessive coding changes in the opposite allele.8, 9, 10
A third possible mechanism exists for deletions or duplications with breakpoints disrupting two different genes. Where the two neighboring genes are encoded on the same chromosomal strand, such CNVs could potentially result in gene-fusion transcripts. A number of genomic rearrangements that lead to fusion transcripts have already been described in autism and schizophrenia.11, 12, 13 If such transcripts are stable they may be translated into novel proteins with a possibility for deleterious gain-of-function effects.
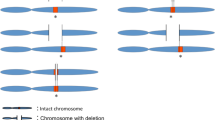
Fusion-proteins have an important role in cancer genetics, most notably in leukemia, and can result from chromosomal translocation (eg BCR-ABL fusion gene on the Philadelphia chromosome). Somatic deletions of chromosome 21 have also been shown to result in TMPRSS2-ERG fusion transcripts in the majority of prostate cancers,14 and the exact configuration of these transcripts appears to correlate with clinical outcome.15 However, to our knowledge, there have been few systematic studies determining the frequency with which germline deletions and duplications can similarly lead to fusion genes/proteins (Figure 1a and b). Therefore, to address the question of how common CNV-induced fusion transcripts are and to assess whether this type of CNV may have a role in ASD susceptibility, we carried out a bioinformatic analysis of existing CNV calls based on published 1M SNP array data.4 Validation of specific instances of such CNVs was then attempted using RT-PCR.

Detection of CNVs that may lead to fusion transcripts. (a) Schematic diagram showing two neighboring genes (blue and green) encoded on the same chromosomal strand. Black arrows show hypothetical position of exonic primers. Below, a deletion leads to a fusion gene whereby, upon transcription, the primers are close enough for efficient RT-PCR amplification to take place. (b) As above, but instead showing a tandem duplication. N.B. As well as the fusion gene, there is one functional copy of both genes on the mutated chromosome. (c) BeadStudio screenshot showing raw Log R Ratio and B allele frequency for a duplication on chromosome 12 in sample 3044.6. The duplication results in an increase to the Log R ratio and deviation in the allelic ratio for heterozygote variants away from the expected 0.5. Breakpoints disrupt different genes, encoded on the same chromosomal strand. SNPs within the duplication are highlighted in red. Region shown is chr12:110 600 000–110 900 000 (NCBI36/hg18).
Materials and methods
CNV calls
CNVs were called as part of a previous AGP study.4 Briefly, DNA from individuals with ASD, parents and controls were genotyped using the Infinium 1M-single SNP microarray (Illumina, San Diego, CA, USA). High confidence CNVs were predicted by intersecting CNV calls from both iPattern and QuantiSNP algorithms (log Bayes Factor >15). Previous analysis showed that validation rates were ∼95% for CNVs identified using this method.4 From these data, rare CNV calls, present at <1% of the combined sample, and greater than 30 kb in size were identified from a total of 996 cases and 1287 controls (all Caucasian). For CNVs of interest, raw SNP data was reviewed manually using BeadStudio version 3.0 (Illumina) (Figure 1c).
Bioinformatic analyses
We used a bioinformatics approach to identify CNVs that show evidence of the breakpoints disrupting two different genes on the same strand. The following steps were applied: because of the relatively high density of SNPs compared to the size of the genes, the identified boundaries of the CNV were considered to be sufficiently exact. Those boundaries were tested against the location of RefSeq genes (NCBI build36/hg18) for overlap. Pairs of genes, transcribed in the same direction, and each containing an extremity of a CNV were flagged and counted. This selection also marked all intermediate genes in the same direction. The procedure was performed independently in the case and control groups. It was therefore possible to compare the number of times a group of genes was affected by a CNV between the two groups.
To determine whether the MAPKAPK5-ACAD10 and POLR1A-REEP1 duplications were ancestral, present on a single haplotype, genotype data for each trio was extracted from BeadStudio. Genotypes were phased for a total of 100 SNPs flanking the duplication (50 on either side or else until the shared haplotype finished). Haplotypes upon which the duplication was observed were then compared.
Quantitative PCR of CNV involving SHANK3
For family 3379, quantitative PCRs were performed in triplicate using the iQ SYBR Green Supermix (BioRad, Hercules, CA, USA), 0.2 μ M of each primer and 20 ng of template DNA in a total reaction volume of 25 μl. Thermocycling and data acquisition was carried out using the iQ5 iCycler (BioRad). Primers sets for SHANK3 targeted intron 3, exon 10, exon 16 and intron 19 are as described.16 A control amplicon in the MAPK1 gene on chromosome 22q11.2 was also included.
Fine mapping the duplication involving REEP1 and POLR1A
Long-range PCR was performed using primers 5′-GATATTGCTGCCCTTCTTGG-3′ and 5′-TGCAAAGAGCCAGCCTAGTT-3′ and the SequalPrep Long PCR kit (Invitrogen, Eugene, OR, USA) according to the manufacturer's suggested protocol. PCR products were purified using exonuclease I (NEB, Ipswich, MA, USA) and shrimp alkaline phosphatase (USB, Cleveland, OH, USA). Sanger sequencing of the junction fragment was then performed using BigDye chemistry (Applied Biosystems, Foster City, CA, USA). The forward PCR primer was sufficiently close to reach the breakpoint (Supplementary Figure 1a).
Fine mapping the duplication involving KIAA0319 and TDP2
Long-range PCR was performed using primers 5′-ACGAGATGTGGCCAAAGTAG-3′ and 5′-ATTCTTGTTCTATTTGGCAGAC-3′ and the BIO-X-ACT long DNA polymerase kit (Bioline, London, UK) according to the manufacturer's suggested protocol. Sequencing was performed as described above, with each PCR primer and also an internal primer (5′-ACCTAATATTGAGTGTTTATGC-3′) that was required to reach the breakpoint (Supplementary Figure 1b).
RNA extraction and cDNA synthesis
EBV-transformed peripheral lymphoblast cell lines (LCLs) were available for a subset of patients. Cells were grown in RPMI 1640 media supplemented with 10% fetal bovine serum (Invitrogen), L-glutamine (final concentration 2 mM) and penicillin (500 U/ml) and streptomycin (5 μg/ml). RNA was extracted using the RNeasy kit (Qiagen, Crawley, UK) and cDNA was synthesized using the QuantiTect reverse transcriptase kit (Qiagen) according to the manufacturer’s suggested protocol, using approximately 1 μg of RNA as template.
RT-PCR and sequencing
PCR primers were designed in exons closest to the CNV breakpoints, following manual inspection of log R ratios and allelic ratio data within BeadStudio. All CNVs tested were duplications; therefore the primers were designed as indicated (Figure 1b; Supplementary Figure 1a-e). For CNVs where the breakpoints were not clear, multiple primer combinations were tested. Primer sequences are listed in Supplementary Table 1.
Thirty five cycles of PCR amplification were performed in a 20-μl volume, with 0.5 μl of cDNA as template, a final Mg2+ concentration of 1.5 mM, each primer at 200 nM and the BIOTAQ DNA polymerase (Bioline). The gene encoding beta-actin (ACTB) was used as a control. PCR products were visualized by UV illumination of 1.8% agarose gels stained with SYBR Safe (Invitrogen). Sequencing was performed as described above.
Expression analysis of the 5′ genes
To test whether the promoters of the genes comprising the 5′ end of the putative fusion transcripts were active in LCLs, cDNA was generated (detailed above). Primers were designed for the 5′ gene of the putative fusion product (Supplementary Table 1) and RT-PCR performed, with ACTB used as a positive control. PCRs were typically carried out using the PCR conditions described above. An annealing temperature of 55 °C was used in all instances except ACTB (56 °C). Expression was determined by the detection of a single band of the predicted size.
The various analysis strategies employed are also summarized in Supplementary Figure 2.
Results
Genome-wide analysis of fusion CNV frequency
In a previous study analyzing 996 individuals with ASD, a total of 2382 rare CNVs present in 889 individuals were observed.4 Similarly, in 1287 control subjects, there were 3096 rare CNVs present in 1146 individuals. From these, we assessed which of these variants had the potential to lead to a fusion transcript. Intersecting CNVs with RefSeq gene coordinates indicated that 134/2382 (5.6%) and 200/3096 (6.5%) of rare CNVs could lead to fusion transcripts in cases and controls, respectively. Reported at the individual level, 122/996 ASD cases harbored at least one rare CNV of this type, compared with 179/1287 control subjects (P=0.89, Fisher’s Exact Test). The frequencies distribution of putative fusion transcript forming CNV was not significantly different in cases compared with controls (P>0.05; Supplementary Figure 3). The synapse is known to be of importance in ASD susceptibility. We therefore compared the frequency between cases and controls of putative fusion transcript forming CNVs involving at least one gene expressed in the human post-synaptic density.17 However, no significant difference was observed (P>0.05).
Despite the lack of a genome-wide increase in burden of potential fusion-gene generating CNVs in ASD, specific instances could still be of potential significance. Therefore, we further examined this class of CNV at single loci.
REEP1-POLR1A fusion gene
Initially, we decided to further examine individual putative fusion-gene generating CNVs based on differences in distribution between cases and controls. The CNV closest to reaching significance was a duplication on chromosome 2. This CNV was present in 3/996 cases (probands from families 13 131, 13 129, 14 284) and 0/1287 controls (P=0.08, Fisher’s Exact Test). Analysis of SNP data indicated that the duplications were on the same haplotype (data not shown) and thus likely to represent an ancient ancestral event. Fine mapping experiments were carried out in family 14 284 that indicated the duplication was a direct tandem repeat involving chr2:86 136 225–86 364 443 (Figure 2a). This rearrangement thus fuses the 3′ end of REEP1 with the 5′ end of POLR1A. Forward and reverse RT-PCR primers were designed in REEP1 and POLR1A (Supplementary Figure 1a) and tested using cDNA from family 14 284 (the one family for whom cDNA was available; in this family the father and proband carry the variant). However, all primer combinations tested failed to generate a PCR product.

Further characterization of CNVs of interest and validation of a fusion transcript. (a) Sequencing electropherogram showing genomic breakpoints for the KIAA0319-TDP2 and POLR1A-REEP1 duplications. (b) Results from RT-PCR analysis of RNA from four cell lines (two probands and two controls). The band at 243 bp indicates that the predicted fusion transcript is present. For each sample a positive and negative RT control was included (+ and − symbols, respectively). (c) Pedigrees showing the segregation pattern of the MAPKAPK5-ACAD10 duplication in families 3044 and 3075. Presence of the fusion transcript duplication is highlighted in red. Arrows indicate the proband. (d) Sequencing electropherogram further validated the MAPAPK5-ACAD10 fusion transcript, with predicted reading frame shown above.
Candidate gene approach
Next, we identified two CNVs as being of potential interest owing to the presence of genes strongly implicated in neurodevelopmental disorders. The first was a 96-kb putative de novo duplication of 22q13 in individual 3379.3 with predicted breakpoints in SHANK3 and RPL23AP82 (Table 1). SHANK3 is a known ASD susceptibility gene.16, 18, 19 However, closer examination of the SNP genotype data indicated that the CNV call was likely to be a false positive. This was confirmed by qPCR analysis of genomic DNA (data not shown).
The second CNV was an inherited duplication on 6p22.2 in Irish subject 13 083.973. This was predicted to fuse the first exon of KIAA0319 with the 3′ end of the neighboring gene TDP2. KIAA0319 is a dyslexia susceptibility gene20, 21, 22 and there is known overlap of susceptibility CNVs between different neurodevelopmental disorders.23, 24 The resolution of the CNV in the 1M SNP data was sufficient to design long-range PCR primers and subsequent PCR confirmed the presence of the CNV as a tandem duplication in the proband and mother. PCR products were sequenced and the duplicated segment identified as chr6:24 728 714–24 766 602 (Figure 2a). Therefore, the CNV would fuse the first noncoding exon of KIAA0319 with exons 4–7 of TDP2. It is unclear whether a viable open reading frame would result from a transcript with this combination of exons. EBV-transformed LCLs were obtained for both individuals carrying the CNV and used to generate cDNA. However, no fusion transcript was detectable by RT-PCR.
Identification of an expressed fusion transcript
None of the previous three CNVs examined above generated a bone fide fusion transcript. However, to confirm that this class of CNV can generate fusion transcripts in our cohort, we screened another subset of putative fusion genes. The 134 rare CNVs with potential to cause fusion transcripts were filtered for those present in cases from the International Molecular Genetic Study of Autism Consortium (IMGSAC) cohort, for which we had direct access to LCLs. This identified 19 CNVs from 18 different families (Table 1). The 1M SNP genotyping results for these CNVs were assessed manually within BeadStudio. A number of these (including the putative duplication involving SHANK3) appeared to be likely false positive CNV calls due to noisy SNP data or gaps in coverage on the 1M array. Therefore, cell lines from four families harboring the three CNVs with the strongest evidence from the genotyping data were tested to identify fusion transcripts. Cell lines for probands were cultured, RNA extracted and used to generate cDNA. Primers were then designed to span between the two genes in the potential fusion transcript.
We were unable to detect transcripts for the CNVs fusing SLC38A8-MBTPS1 or DSG3-DSG4 from the probands of families 3199 and 3309, respectively. A PCR product was obtained for samples from families 3044 and 3075 showing that the ∼130-kb duplication on chromosome 12, joining the 5′ end of MAPKAPK5 and the 3′ end of ACAD10, does lead to a bone fide fusion transcript (Figure 2b). Analysis of other family members confirmed that this CNV had been inherited by the probands’ affected sibling in both families, as well as one unaffected brother in family 3044 (Figure 2c). Sequencing of the PCR products showed the transcript runs from exon 7 of MAPKAPK5 to exon 13 of ACAD10. The ACAD10 portion of this novel transcript is out of frame and predicted to lead to 13 new amino acids followed by a stop codon (Figure 2d). Analysis of the SNPs flanking the duplication in all three IMGSAC families showed the duplication bearing haplotype to be identical, indicating the CNV was ancestral (Supplementary Table 2). Overall this CNV was seen in 5/996 (0.5%) of cases and 5/1287 (0.4%) of controls.
Confirmation of gene expression in lymphoblasts
One potential explanation for the lack of observation of fusion transcripts was that the promoter of the 5′ gene is simply not active in LCLs. Therefore, we extracted RNA from control LCLs and subsequently generated cDNA. Transcripts were detected for POLR1A, KIAA0319 and DSG3. However, we were unable to detect expression of SLC38A8. Thus, in the majority of cases examined, if a fusion transcript were generated, we would expect to observe it using the methods employed here.
Discussion
In this study, we have assessed whether CNVs that lead to fusion transcripts may have a role in susceptibility to ASD. To do this we integrated CNV and RefSeq gene coordinates, looking for CNVs where the breakpoints intersect two different genes that are transcribed in the same direction. Our analysis indicates that there is no increased burden of this type of CNV in 996 cases compared with 1287 controls.
The sample size currently available gives our study limited power to detect association of CNVs at specific loci. For instance, in our analysis, the CNV closest to reaching significance involved the REEP1 and POLR1A genes and reached P=0.08. POLR1A and REEP1 encode polymerase (RNA) I polypeptide A and a mitochondrial receptor accessory protein, respectively. Interestingly, dominant mutations of REEP1 lead to autosomal-dominant hereditary spastic paraplegia, and a recent study described a similar duplication involving exons 2–7 as part of the mutational spectrum of this gene.25 It is possible that the loss of POLR1A 3′-regulatory elements in the fusion gene may lead to a loss of expression in LCLs, but that expression might still occur in other cell types. However, no other tissue types were available from the families to test this hypothesis. Therefore, this CNV may still potentially have a role in ASD susceptibility through our hypothesized mechanism.
Investigation of two candidate CNVs based on the previous identification of the genes involved in neurodevelopmental disorders indicated that neither were likely to be involved in ASD susceptibility through our hypothesized mechanism. The putative de novo duplication on chromosome 22q, involving SHANK3 and RPL23AP82, was assessed by qPCR and shown to be a false positive. This finding illustrates one significant limitation of this study – that although two separate CNV detection algorithms were used to improve the confidence of CNV calling,4 upon closer examination, a proportion of the potentially fusion transcript forming CNV calls appeared to be imprecise in terms of breakpoints or else are false positives. Comparative studies have shown that there is still a surprising amount of variation between CNV calling algorithms, even when using the same raw data.26 Further work to improve CNV calling algorithms would in turn improve accuracy of predicting CNVs that may lead to fusion transcripts. In contrast, the duplication involving KIAA0319 (a susceptibility gene for dyslexia) was validated using genomic DNA, although no fusion transcript was identified.
In this study, a fusion transcript involving the MAPKAPK5 and ACAD10 genes was validated in two multiplex families. In both families tested, this chromosome 12 duplication and associated transcript were present in the probands’ affected sibling, as well as an unaffected brother in family 3044 (Figure 2c). However, sequencing indicated that the transcript had a premature stop codon and so may be degraded by nonsense-mediated decay. Within the whole AGP study, this variant was seen at a similar frequency in cases and controls. Thus, it is unlikely to have relevance to ASD susceptibility. It is interesting to note that ALDH2 (encoding the aldehyde dehydrogenase 2 enzyme) is fully contained within this duplication. This gene is known to influence alcohol sensitivity27 and risk for esophageal cancer,28 and so it may be that this duplication confers some protection against these two conditions. The ancestral nature of this duplication suggests it is likely to be imputable in GWAS analysis, making this hypotheses easily testable.
One potential weakness in our attempts to confirm the presence or absence of fusion transcripts is the use of cDNA generated from LCLs, as opposed to cell types more directly related to ASD. However, work by Baron et al29 has suggested that LCLs are suitable substitutes and several studies have used them to investigate ASD risk factors.30, 31
There are a number of reasons why 80% of the CNVs tested may not have resulted in detectable fusion transcripts. As discussed earlier, the gene making up the 5′ end of fusion product may not be expressed in LCLs. Our RT-PCR analyses appear to rule this possibility out for three of the CNVs (although it must be noted that large CNVs might alter chromatin structure). Second, it may be that the loss of 3′ control elements from the 5′ fused gene may alter expression, such that the fused product is not seen in LCLs. This may be the case with the POLR1A-REEP1 and KIAA0319-TDP2 CNVs. Third, it could be that fusion transcripts are expressed, but at levels below the sensitivity of the RT-PCR method used here. Fourth, the neighboring splice donor and acceptor sites may simply be incompatible and so may result either in unexpected cryptic transcripts (a possibility that could be addressed in future studies by the use of 3′-RACE or RNAseq) or else in a total absence of fusion transcript. Fifth, the resolution of CNV breakpoints may not have been sufficiently accurate to enable suitable primers to be designed. Finally, as all these CNVs happened to be duplications, we could not always be sure that the duplicated DNA was not present at a different genomic locus. The exception to the last two points are the POLR1A-REEP1 and KIAA0319-TDP duplications, where fine-mapping confirmed the CNVs to be in a direct tandem orientation (Figure 2a).
In conclusion, our data do not lend support to the hypothesis that gain-of-function fusion genes are a common mechanism for ASD susceptibility. However, larger data sets are required to better assess individual loci. Our characterization of a MAPAPK5-ACAD10 fusion transcript is consistent with other studies showing that rare germline CNVs can lead to fusion transcripts. Therefore, fusion genes and gain-of-function mechanisms should still be considered in future studies of genomic imbalance and disease susceptibility.
References
Redon R, Ishikawa S, Fitch KR et al: Global variation in copy number in the human genome. Nature 2006; 444: 444–454.
Perry GH, Dominy NJ, Claw KG et al: Diet and the evolution of human amylase gene copy number variation. Nat Genet 2007; 39: 1256–1260.
Craddock N, Hurles ME, Cardin N et al: Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010; 464: 713–720.
Pinto D, Pagnamenta AT, Klei L et al: Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Morrow EM, Yoo SY, Flavell SW et al: Identifying autism loci and genes by tracing recent shared ancestry. Science 2008; 321: 218–223.
Stranger BE, Forrest MS, Dunning M et al: Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007; 315: 848–853.
Henrichsen CN, Vinckenbosch N, Zollner S et al: Segmental copy number variation shapes tissue transcriptomes. Nat Genet 2009; 41: 424–429.
Flipsen-ten Berg K, van Hasselt PM, Eleveld MJ. et al: Unmasking of a hemizygous WFS1 gene mutation by a chromosome 4p deletion of 8.3 Mb in a patient with Wolf-Hirschhorn syndrome. Eur J Hum Genet 2007; 15: 1132–1138.
Pagnamenta AT, Khan H, Walker S et al: Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet 2011; 48: 48–54.
Vorstman JA, van Daalen E, Jalali GR et al: A double hit implicates DIAPH3 as an autism risk gene. Mol Psychiatry 2011; 16: 442–451.
Pagnamenta AT, Bacchelli E, de Jonge MV et al: Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psych 2010; 68: 320–328.
Walsh T, McClellan JM, McCarthy SE et al: Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008; 320: 539–543.
Zhou X, Chen Q, Schaukowitch K, Kelsoe JR, Geyer MA : Insoluble DISC1-Boymaw fusion proteins generated by DISC1 translocation. Mol Psych 2010; 15: 669–672.
Tomlins SA, Rhodes DR, Perner S et al: Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005; 310: 644–648.
Wang J, Cai Y, Ren C, Ittmann M : Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res 2006; 66: 8347–8351.
Bonaglia MC, Giorda R, Mani E et al: Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet 2006; 43: 822–828.
Bayés A, Lagemaat LN, Collins MO et al: Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 2011; 14: 19–21.
Durand CM, Betancur C, Boeckers TM et al: Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39: 25–27.
Moessner R, Marshall CR, Sutcliffe JS et al: Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet 2007; 81: 1289–1297.
Harold D, Paracchini S, Scerri T et al: Further evidence that the KIAA0319 gene confers susceptibility to developmental dyslexia. Mol Psych 2006; 11: 1085–1091.
Paracchini S, Steer CD, Buckingham LL et al: Association of the KIAA0319 dyslexia susceptibility gene with reading skills in the general population. Am J Psych 2008; 165: 1576–1584.
Paracchini S, Thomas A, Castro S et al: The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet 2006; 15: 1659–1666.
Levinson DF, Duan J, Oh S et al: Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psych 2011; 168: 302–316.
Williams NM, Zaharieva I, Martin A et al: Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Beetz C, Schule R, Deconinck T et al: REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008; 131: 1078–1086.
Pinto D, Darvishi K, Shi X et al: Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotech 2011; 29: 512–520.
Crabb DW : Biological markers for increased risk of alcoholism and for quantitation of alcohol consumption. J Clin Invest 1990; 85: 311–315.
Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A : The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med 2009; 6: e50.
Baron CA, Liu SY, Hicks C, Gregg JP : Utilization of lymphoblastoid cell lines as a system for the molecular modeling of autism. J Autism Dev Disord 2006; 36: 973–982.
Hu VW, Nguyen A, Kim KS et al: Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: altered pathways in neuronal development and steroid biosynthesis. PLoS One 2009; 4: e5775.
Yasuda Y, Hashimoto R, Yamamori H et al: Gene expression analysis in lymphoblasts derived from patients with autism spectrum disorder. Mol Autism 2011; 2: 9.
Acknowledgements
We gratefully acknowledge the families participating in the study, Dalila Pinto and members of the international Autism Genome Project consortium for sharing data, ideas and comments on the manuscript. Funding was from Autism Speaks and a Wellcome Trust core award 090532/Z/09/Z. ATP is currently supported by the NIHR Biomedical Research Centre. We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. The Autism Genetic Resource Exchange is a program of Autism Speaks and is supported, in part, by Grant 1U24MH081810 from the National Institute of Mental Health to Clara M Lajonchere (PI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Holt, R., Sykes, N., Conceição, I. et al. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur J Hum Genet 20, 1141–1147 (2012). https://doi.org/10.1038/ejhg.2012.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.73
Keywords
This article is cited by
-
Characterization of a novel loss-of-function variant in TDP2 in two adult patients with spinocerebellar ataxia autosomal recessive 23 (SCAR23)
Journal of Human Genetics (2020)
-
Pediatric asthma and autism—genomic perspectives
Clinical and Translational Medicine (2015)
