
Abstract
For many complex diseases, quantitative traits contain more information than dichotomous traits. One of the approaches used to analyse these traits in family-based association studies is the quantitative transmission disequilibrium test (QTDT). The QTDT is a regression-based approach that models simultaneously linkage and association. It splits up the association effect in a between- and a within-family genetic component to adjust and test for population stratification and includes a variance components method to model linkage. We extend this approach to detect gene–gene interactions between two unlinked QTLs by adjusting the definition of the between- and within-family component and the variance components included in the model. We simulate data to investigate the influence of the epistasis model, linkage disequilibrium patterns between the markers and the QTLs, and allele frequencies on the power and type I error rates of the approach. Results show that for some of the investigated settings, power gains are obtained in comparison with FAM-MDR. We conclude that our approach shows promising results for candidate-gene studies where too few markers are available to correct for population stratification using standard methods (for example EIGENSTRAT). The proposed method is applied to real-life data on hypertension from the FLEMENGHO study.
Similar content being viewed by others


Introduction
In order to disentangle the genetic basis of complex diseases, the initial focus is usually on detecting associations between single SNPs and a specific phenotype. Over time, it has become clear that only partial information can be obtained by performing single marker analyses, for example, due to the presence of epistasis or gene–gene interactions. To test for gene–gene interactions in family data, both non-parametric methods (MDR-PDT1) and parametric methods (FITF2 and FAM-MDR3) exist. Some difficulties are encountered when opting for a family design, one of which involves data collection. However, family-based studies offer several advantages, including the possibility to use tests that are robust to population stratification. The aforementioned methods are not robust against population stratification. Also, MDR-PDT and FAM-MDR only test for association and do not automatically account for previously detected linkage signals. It is suggested that this can increase type I error rates in specific settings, for example, in the case of a rare private allele.4
The QTDT5 is a mixed model approach to test for association in the presence of linkage in the family data for quantitative traits. In this paper, we propose an extension of the QTDT approach (noted by epiQTDT) that tests for gene–gene interactions in the presence of linkage and at the same time is robust against population stratification. It is applicable to pedigrees of any size. We perform an extensive simulation study and show estimated power and type I error rates in nuclear families with two offspring and with and without missingness in all parental genotypes. Markers have differing minor allele frequencies (MAF) and we consider several degrees of linkage disequilibrium (LD) between the markers and the quantitative trait loci (QTLs). We also apply the epiQTDT method to a real-life data set from the Flemish Study on Environment, Genes and Health Outcomes (FLEMENGHO) on hypertension.
Materials and methods
The model
Assume that two unlinked additive QTLs are influencing a trait Y. First, we consider unrelated individuals and denote for an individual i the recoded genotypes at the QTLs by G1i and G2i and the trait value by Yi. We use the additive coding (−1, 0, 1) for the recoded genotypes. In the presence of a gene–gene interaction between the two QTLs, we can model the mean trait value by:

This model assumes that the individuals are independent and that the trait values are normally distributed conditionally on the recoded genotypes. For family designs, the assumption that the individuals are independent is not valid. The variance–covariance matrix of the trait values of individuals j and k from the same family i has the following expression:

In (2), φijk is the coefficient of relationship between individual j and k (twice the kinship coefficient), πijk1 is the proportion of alleles-shared IBD between individuals j and k at locus 1 (analogous for πijk2) and ⊗ is the Hadamard product (element-wise multiplication) between two matrices. We use the notation σa12, σa22 and σaa2 for the additive genetic variance of the trait explained by locus 1, locus 2 and the interaction between the two loci, respectively. Furthermore in (2), σp2 is the polygenic variance and σe2 is the non-shared environmental variance. This variance components model is based on Mitchell et al.6
The mixed model that results from combining the expressions in (1) and (2) can be fitted using likelihood theory (Restricted Maximum Likelihood (REML) principles). It is important to realise that the model for the mean trait value still suffers from phenomena like population stratification or population admixture, which can lead to spurious associations.5 To deal with this issue when targeting one QTL, we split the genetic effects into two orthogonal components: a within- and a between-family component.5 Consider for instance a set of N nuclear families and a locus Q. For the jth individual of the ith family, Gij is the recoded genotype at Q, GMi and GFi are the recoded genotypes at Q for the male and female founder, and Si is the set of full siblings. Further define Gi as the set of genotyped individuals in family i. For the founders j of family i, the between-family component bij is defined as the recoded genotype Gij. The between-family component bi for a non-founder individual j of family i is then defined as followed:

Hence, when all parental genotypes are known, the between-family component is defined as the expected genotype for offspring j of family i conditional on the parental genotypes.
The within-family component wij for individual j of family i is simply defined as the difference between the recoded genotype and the between-family component:

The advantage of the split-up of the genotype information in two components is that testing the within-family component for association is not influenced by population stratification. This previously introduced decomposition can easily be extended to larger pedigrees.7
When considering N nuclear families and using the decomposition for both QTL 1 and 2, model (1) can be rephrased as:

where w1, w2, b1 and b1 are the within-family and between-family components for QTL 1 and 2, and w12 and b12 are the within-family and between-family component of the product of the two recoded genotypes at locus 1 and 2. Similar to (3), the between-family component of this product is defined as the expected value of this product, given the parental genotypes. As the two QTLs under study are assumed to be unlinked, this is the product of b1 and b2. The within-family component w12 is then defined as the difference between the product of the two recoded genotypes and the between-family component. Again, those two components are orthogonal. In the Supplementary Material (Supplementary Appendix A), we show that the estimated within-family coefficient α̂12 of the gene–gene interaction is an unbiased estimator for the interaction effect and calculate formulae for the bias of the estimated between-family coefficient β̂12 of the gene–gene interaction in the presence of population stratification. As we are looking at related individuals in model (4), we model the covariance between two individuals again according to (2).
Amos8 observed that, when handling markers instead of unknown QTLs, the additive variances in the variance–covariance structure (2) are influenced by the recombination fractions between the markers and the QTLs. Therefore, we will assume that each of the two markers is perfectly linked to one of the QTLs. Under these assumptions, the model constructed above can still be used when testing markers instead of QTLs for association. If we test for epistasis between the two markers, we consequently test whether α12=0 in model (4) and refer to this test as ‘epiQTDT’. EpiQTDT tests for gene–gene interactions conditional on main effects. We also construct a joint test of the main effects and interaction effect by testing simultaneously.
FAM-MDR
We compare the performance of epiQTDT to FAM-MDR,3 a multifactor dimensionality reduction (MDR) method that combines the features of the genome-wide rapid association using mixed model and regression (GRAMMAR9) approach with model based-MDR (MB-MDR10, 11). First, we fit a polygenic model, specifying a mean model containing an intercept and possibly covariates. Second, we construct residuals based on this model, which are assumed to be free of all correlations due to polygenic effects. Third, we apply MB-MDR using these residuals as a new ‘family-corrected’ outcome. Step down maxT12 is used to correct for multiple testing.
To obtain results that are comparable to the epiQTDT approach, we account for main effects in the MB-MDR method.11 Both epiQTDT and FAM-MDR will thus test for gene–gene interactions in family data conditional on any main effects. We note that there are some differences between the two methods: epiQTDT accounts for linkage signals and corrects for population stratification, where FAM-MDR does not.
Testing for population stratification
In Supplementary Appendix A, we show the following properties for the estimates of the coefficients in model (4) (for a simple linear regression):

where a1, a2 and a12 are the coefficients of the regression model (1). The exact formulae for the bias can be found in Supplementary Appendix A as well. We can conclude from these properties that all within-family effects are different from the between-family effects when population stratification is present. Therefore, to test for population stratification, we use a joint test to test whether (α1, α2, α12) is equal to (β1, β2, β12) or not in model (4). The properties of this test are illustrated in the simulation study.
Data simulation
To assess results for type I error rates and power, we simulate genetic data for two unlinked QTLs and two markers in LD with the QTLs. We extend the LD between the markers and the QTLs from D′=0 to 1 by 0.1. D′=0 represents the situation for no LD between markers and QTLs and thus no association between the trait and the marker genotypes. We can assess type I error rates in these simulated settings. The MAFs of the QTLs and the markers are assumed to be equal and to vary from 0.1 to 0.3 and 0.5. For each setting, we simulate 1000 data sets and 1020 offspring (in nuclear families). We simulate data with nuclear families containing two offspring in the absence (Scenario 1) and presence (Scenario 2) of population stratification.
Scenario 1: no population stratification
In the absence of population stratification, we simulate three settings. In the first setting, the additive variance of the trait explained by each of the QTLs is 1, and the additive variance of the trait explained by the gene–gene interaction is 3. We add a (non-shared) environmental variance of 65 and a polygenic variance of 30. These choices are motivated by the work of Abecasis.5 The heritability of the trait is in this setting thus 0.05. For the second setting, we double the additive genetic variances of the trait explained by the QTLs and the gene–gene interaction, leading to a heritability of the trait of 0.095. The third setting sets the additive variance of the trait explained by each of the QTLs equal to 0 and the additive variance of the trait explained by the gene–gene interaction equal to 5. This setting includes no explicit main effects of the QTLs but we know that simulating data according to this scenario induces weak main effects.13 The heritability of the trait for the third setting is 0.05. We can compare setting 1 and 3 to compare a setting with explicit main effects with a setting without explicit main effects. The heritability in both settings is the same.
The simulation of the trait values according to the specified variance decompositions is based on drawing values from a multivariate normal distribution; details about this can be found in Supplementary Appendix B. We note that in the above-mentioned simulated settings, the additive effect of the gene–gene interaction is larger than the main effects of the QTLs.
For these settings, we compare power and type I errors for epiQTDT, the joint epiQTDT and FAM-MDR. We also investigate data where all genetic information for the parents is missing to see how well epiQTDT copes with missing parental genotypes. In this case, we remove all other available information from the parents before fitting the mixed models of the epiQTDT and joint epiQTDT.
Scenario 2: population stratification
In the presence of population stratification, we define the regression coefficients in model (1) to simulate the trait values. This allows us to measure the bias that is induced in the between-family coefficient estimates and to check whether the within-family association estimates are truly unbiased. We simulate two population strata, each containing an equal number of nuclear families. For every simulated nuclear family, both founders are drawn from the same stratum. We do not consider population admixture. The two unlinked QTLs and markers have MAF 0.5 in the first stratum and have MAFs 0.1, 0.3 or 0.4 in the second stratum. The population mean of the trait in the first stratum is 10 and in the second stratum, the population mean is 1.
To simulate the trait values, we consider two different settings. First, we assume that a1=a2=1.5 and a12=2 in model (1) (setting 4). For MAFs 0.1, 0.3, 0.4 and 0.5, the heritabilities of trait are, respectively, 0.015, 0.067, 0.097 and 0.12. We note that this setting represents a simulation where the main effects of the QTLs are larger than the interaction effect of the QTLs. In a second setting, we set a1=a2=−1.5 and a12=3 in model (1) (setting 5). This results in a setting where the additive genetic variance explained by both QTLs is smaller than the additive epistatic genetic variance. For MAFs 0.1, 0.3, 0.4 and 0.5, the corresponding heritabilities of the trait are 0.0061, 0.017, 0.029 and 0.045.
For both simulation settings, we compare power and type I error rates of epiQTDT, joint epiQTDT and FAM-MDR, and measure the bias in the estimates of the between-family component effects. We also investigate the power of our test for population stratification.
Results
Scenario 1: no population stratification
Type I error rates for some of the settings are available in Table 1. We conclude that, when no population stratification is present, the type I error rates are in general close to 0.05 and that both epiQTDT and FAM-MDR tend to be conservative.
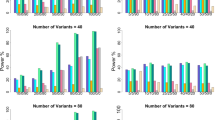
Figure 1 displays the power results for simulation setting 1. Setting 1 includes main effects for both QTLs and has a trait with a heritability of 0.05. We conclude that the joint epiQTDT has the best performance. As the joint epiQTDT is the only considered method that tests for main effects and an interaction effect simultaneously, this is in line with the expectations. Both epiQTDT and FAM-MDR try to detect epistasis conditional on any main effects. In most settings where the parental genotypes are not missing, FAM-MDR outperforms epiQTDT. Only for low values of D′, epiQTDT has equal or more power than FAM-MDR. When the parental genotypes are all missing, the epiQTDT shows better results. In summary, epiQTDT and FAM-MDR have comparable power in setting 1. For MAFs 0.5 and 0.3, epiQTDT does slightly better; for MAF 0.1, the opposite is true.

Power and type I error rates (simulation setting 1: h2=0.05, no population stratification, main effects present) with and without (all) parental genotypes missing as a function of the degree of LD between the markers and QTLs. D′=0 results in type I error rates.
Figure 2 displays the power results for simulation setting 2. Simulation setting 2 resembles setting 1, but includes a trait with a heritability close to 0.1. In Figure 2, we observe similar patterns for setting 2 than for setting 1. The power for all methods is higher, due to the larger heritability of the trait.

Power and type I error rates (simulation setting 2: h2≈0.1, no population stratification, main effects present) with and without (all) parental genotypes missing as a function of the degree of LD between the markers and QTLs. D′=0 results in type I error rates.
Finally, Figure 3 shows the power results for simulation setting 3, where the trait has a heritability of 0.05 (as in setting 1). Setting 3 does not include explicit main effects of the QTLs. We observe that in this case, the joint epiQTDT performs poorly, due to the absence of explicit main effects of the QTLs. For epiQTDT and FAM-MDR, the same observations as for Figures 1 and 2 hold.

Power and type I error rates (simulation setting 3: h2=0.05, no population stratification, no explicit main effects) with and without (all) parental genotypes missing as a function of the degree of LD between the markers and QTLs. D′=0 results in type I error rates.
Scenario 2: population stratification
Table 1 contains the type I error rates for FAM-MDR and the tests for the within-family and between-family component of the gene–gene interaction. The elevated type I errors of the test for the between-family component are only clear when we mix MAF (for all markers and QTLs) 0.1 in one of the strata with 0.5 in the other stratum. For the other MAFs, the test for the between-family component of the gene–gene interaction performs quite well. However, in these settings, the test of the between-family component of the main effects in the model have elevated type I errors (results not shown). For FAM-MDR (that does not correct for population stratification) we observe the same trends.
Figure 4 shows the power results for simulation setting 4 and 5 in the presence of population stratification. In simulation setting 4, the additive genetic variance of the trait due to both QTLs is larger than the additive genetic epistatic variance. In simulation setting 5, the additive genetic epistatic variance of the trait is larger than the additive genetic variance of the trait explained by both QTLs. In Figure 4, we have added some more information: power results for the test for population stratification and power results for the test of the between-family component of the gene–gene interaction (in addition to the power of the test of the within-family component, so far referred to as epiQTDT). When the population stratification is severe (mix of MAFs 0.1/0.5), then we clearly see in Figure 4 that the test for the between-family component does not perform well (for both settings). When the population stratification is less severe (mix of MAFs 0.4/0.5), the power of the test for the within-family and between-family component is more alike. When comparing the epiQTDT, joint epiQTDT and FAM-MDR, we notice again that the joint epiQTDT has the best performance, due to the large main effects of the QTLs that are present in setting 4 and 5. When comparing FAM-MDR and epiQTDT, we conclude now that epiQTDT outperforms FAM-MDR (in both settings). The difference in performance is larger when the population stratification is more severe. The inflation of the type I error rate of FAM-MDR is more prominent in case of severe population stratification. In addition, we observe in Figure 4 that the power for the test for population stratification remains constant for varying values of D′ and decreases when the population stratification is less severe.

Power and type I error rates (simulation setting 4: population stratification, main effects > interaction effect and simulation setting 5: population stratification, main effects < interaction effect) as a function of the degree of LD between the markers and the QTLs. D′=0 results in type I error rates.
Finally, in Table 2 we show some results about the bias of the estimates for the between-family components included in model (4). We can clearly see that the estimates for all within-family components are unbiased, whereas the estimates for the between-family components are biased (when D′=1 and the markers coincide with the QTLs). Table 2 also contains power results for the test for population stratification. It shows that our proposed test has adequate power to detect (severe) population stratification.
We note that detailed results of all simulations can be found in Supplementary Appendix C.
Application to hypertension data
To illustrate the performance of epiQTDT on a real-life data set, we use a hypertension data set from the FLEMENGHO study. The FLEMENGHO study is a collection of surveys with data on families in an area of Northern Belgium between 1985 and 1999 (7501 subjects). We refer to Staessen et al14 for more details about the study.
In Staessen et al14 and Li et al15, gene–gene interactions between markers from the angiotensin-converting enzyme gene (ACE, chromosome 17), the α-adducin gene (ADD1, chromosome 4) and the aldosterone synthase gene (AS, chromosome 8) are published. The purpose of our analysis is to identify these gene–gene interactions with our new methodology and to investigate whether novel interactions can be identified.
The genetic data from the FLEMENGHO study contain one marker on each of these three genes (denoted by G_AS, G_ACE and G_ADD1). The three markers are in Hardy–Weinberg equilibrium (HWE) and have an MAF of 0.43 (G_AS), 0.49 (G_ACE) and 0.23 (G_ADD1). The observed outcomes are diastolic blood pressure (dbp) and systolic blood pressure (sbp). Additional information is available on age, sex, bmi and trt_ht (binary variable that indicates whether the subject is under treatment for hypertension or not). Data preparation for analysis includes Mendelian transmission checks. In addition, we remove families where no genotype information is available for any of the family members and eliminate families with one or two individuals only. The total number of retained subjects is 5714. This data reduction does not influence our analysis, as the removed subjects do not contribute to our test statistic (within-family component is 0). The data reduction also speeds up the calculation of the IBD estimates. To construct the IBD estimates, we use MERLIN16 and SIMWALK17 in families where MERLIN was not able to estimate the IBD probabilities. We obtain the IBD estimates independently (single-point estimation) as the three genes are located on different chromosomes.
We test the three proposed gene–gene interactions with dbp as outcome using epiQTDT and FAM-MDR. For epiQTDT, we perform two tests: the first test is the application of epiQTDT using a mixed model without covariates; the second test is based on a mixed model where covariates are included. As there are a lot of missing data, we apply ‘forward model building’ (with dbp as outcome) using the four covariates to obtain an optimal model for the covariates. This leads to the following model:

For the second epiQTDT analysis, we add the within-family and between-family component of the main effects and gene–gene interaction to this model and apply the test for the within-family component of the gene–gene interaction. For both analyses, we use a Bonferroni correction to correct for multiple testing.
For FAM-MDR, we also apply two different tests. In a first test, we do not correct for covariates and apply FAM-MDR as before (and thus again conditioning on main effects). In a second test, we correct for covariates in the GRAMMAR procedure using model (5).
The results for the epiQTDT and FAM-MDR are shown in Table 3. For FAM-MDR, we observe a significant gene–gene interaction between G_AS and G_ADD1 (P-value: 0.024) when correcting for covariates. Unfortunately, we do not know how reliable this result is, as the GRAMMAR method did not converge when correcting for covariates. We note that for FAM-MDR, all P-values are corrected for multiple testing.
For the epiQTDT analysis, we notice two marginally significant results: the gene–gene interaction between G_AS and G_ACE (P-value: 0.072 in the model with covariates) and the gene–gene interaction between G_ACE and G_ADD1 (P-value: 0.087 in the model without covariates). These results look promising but have to be interpreted with care. For both detected gene–gene interactions, the results are not consistent when looking at the models with and without covariates. We detect a gene–gene interaction between G_ACE and G_ADD1 when we ignore covariates in the model. If we add covariates, the gene–gene interaction disappears. A reason for this phenomenon could be a gene–environment interaction between some of the covariates and markers. If we investigate this further, we indeed find a three-way interaction between G_ACE, G_ADD1 and sex (P-value: 0.0404). The opposite is observed for the gene–gene interaction between G_AS and G_ACE. This interaction is not significant in the model without covariates but becomes marginally significant after inclusion of the covariates in the model (5).
Discussion
In this paper, we propose a mixed model approach epiQTDT that tests for gene–gene interactions in family data in the presence of linkage while controlling for population stratification. It is applicable to any type of family structure and can adjust for covariates.
In an extensive simulation study, we show that epiQTDT has good power for traits with high and low heritabilities. When no population stratification is present, we conclude from our simulation study that FAM-MDR outperforms epiQTDT for most of the considered settings. Only for low values of D′ between the markers and the QTLs, small power gains of epiQTDT are established. In this setting, the power of epiQTDT is reduced as we correct for population stratification that is not present. The split-up in within-family and between-family information is not necessary. The power of epiQTDT is reduced in this case because we split up the genotypic information. When all parental genotypes are missing, epiQTDT and FAM-MDR show comparable power results.
When comparing the joint epiQTDT to epiQTDT and FAM-MDR in case of no population stratification, we observe that the joint epiQTDT shows better results for simulation settings 1 and 2, where both QTLs have a main and interaction effect on the trait (Figures 1 and 2). The joint epiQTDT is able to detect these main effects. EpiQTDT and FAM-MDR only test for the interaction effect, thus have less power in these settings. If the two QTLs only have a weak main effect on the trait (simulation setting 3), epiQTDT and FAM-MDR are more powerful than the joint epiQTDT. When all parental genotypes are missing, the same trends are observed.
Finally, in Figure 4, we show that in the presence of population stratification, epiQTDT outperforms FAM-MDR. The epiQTDT and joint epiQTDT correctly address the population stratification and have better power results than FAM-MDR.
The type I error rates of the epiQTDT and joint epiQTDT are always close to 0.05 in the considered settings. We show that the test and estimated effect of the within-family component of the gene–gene interaction is unbiased in the presence of population stratification (Tables 1 and 2). Several methods exist to test or control for population stratification (eg, EIGENSTRAT18) but usually require a lot (>1000) of markers. If too few markers are available to use these methods, epiQTDT offers a nice alternative.
Although the split up of the family information in a within-family and between-family component guaranties unbiased results in the presence of population stratification, there can still be a problem in the covariance model. The IBD estimates can be biased by population stratification when founder genotype data are missing.19 Another problem in the covariance model is the use of the Hadamard product of the IBD estimate matrices of the two loci. The product is only valid when the two QTLs are unlinked (ie, on different chromosomes) or when there is at least one informative marker between the QTLs in case the latter are linked.20 At this point, it is not clear what the effect of these issues on the performance of epiQTDT is, although we do not observe elevated type I error rates in our simulated settings (eg, in the presence of population stratification). We suspect that the influence will be minor, as we are only correcting for linkage and not explicitly testing for it.
Another important assumption of epiQTDT is the multivariate normality assumption of the error terms in the applied mixed model. If this assumption does not hold, conclusions can be biased. Several solutions exist to solve this problem: for example, a permutation-based solution,5 and in Diao and Lin21 a method is proposed that uses an unspecified transformation function that is empirically estimated from the data.
We conclude that epiQTDT shows promising results to detect gene–gene interactions in the presence of linkage in particular in (candidate–gene) studies where too few markers are available to correct for population stratification using standard methods. Supplementary Information is available at the European Journal of Human Genetics’ website.
References
Martin ER, Ritchie MD, Hahn L, Kang S, Moore JH : A novel method to identify gene-gene effects in nuclear families: the MDR-PDT. Genet Epidemiol 2006; 30: 111–123.
Millstein J, Siegmund KD, Conti DV, Gauderman WJ : Identifying susceptibility genes by using joint tests of association and linkage and accounting for epistasis. BMC Genet 2005; 6 (Suppl 1): S147.
Cattaert T, Urrea V, Naj AC et al: FAM-MDR: a flexible family-based multifactor dimensionality reduction technique to detect epistasis using related individuals. PLoS ONE 2010; 5: e10304.
Kent Jr JW, Dyer TD, Goring HH, Blangero J : Type I error rates in association versus joint linkage/association tests in related individuals. Genet Epidemiol 2007; 31: 173–177.
Abecasis GR, Cardon LR, Cookson WO : A general test of association for quantitative traits in nuclear families. Am J Hum Genet 2000; 66: 279–292.
Mitchell BD, Ghosh S, Schneider JL, Birznieks G, Blangero J : Power of variance component linkage analysis to detect epistasis. Genet Epidemiol 1997; 14: 1017–1022.
Abecasis GR, Cookson WO, Cardon LR : Pedigree tests of transmission disequilibrium. Eur J Hum Genet 2000; 8: 545–551.
Amos CI : Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet 1994; 54: 535–543.
Aulchenko YS, de Koning DJ, Haley C : Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics 2007; 177: 577–585.
Calle ML, Urrea V, Vellalta G, Malats N, Steen KV : Improving strategies for detecting genetic patterns of disease susceptibility in association studies. Stat Med 2008; 27: 6532–6546.
Mahachie John JM, Van Lishout F, Gusareva ES, Van Steen K : Lower-order effects adjustment in quantitative traits model-based multifactor dimensionality reduction. Eur J Hum Genet 2011, (In press).
Westfall PH, Y S (ed): Resampling-Based Multiple Testing. New York: John Wiley and Sons, 1993.
Marchini J, Donnelly P, Cardon LR : Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat Genet 2005; 37: 413–417.
Staessen JA, Wang JG, Brand E et al: Effects of three candidate genes on prevalence and incidence of hypertension in a Caucasian population. J Hypertens 2001; 19: 1349–1358.
Li Y, Zagato L, Kuznetsova T et al: Angiotensin-converting enzyme I/D and alpha-adducin Gly460Trp polymorphisms - from angiotensin-converting enzyme activity to cardiovascular outcome. Hypertension 2007; 49: 1291–1297.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Wang T, Elston RC : The bias introduced by population stratification in IBD based linkage analysis. Hum Hered 2005; 60: 134–142.
Ronnegard L, Pong-Wong R, Carlborg O : Defining the assumptions underlying modeling of epistatic QTL using variance component methods. J Hered 2008; 99: 421–425.
Diao G, Lin DY : Improving the power of association tests for quantitative traits in family studies. Genet Epidemiol 2006; 30: 301–313.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
De Lobel, L., Thijs, L., Kouznetsova, T. et al. A family-based association test to detect gene–gene interactions in the presence of linkage. Eur J Hum Genet 20, 973–980 (2012). https://doi.org/10.1038/ejhg.2012.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.45
