
Abstract
Recent reports suggest that somatic structural changes occur in the human genome, but how these genomic alterations might contribute to disease is unknown. Using samples collected as part of the International Schizophrenia Consortium (schizophrenia, n=3518; control, n=4238) recruited across multiple university research centers, we assessed single-nucleotide polymorphism genotyping arrays for evidence of chromosomal anomalies. Data from genotyping arrays on each individual were processed using Birdsuite and analyzed with PLINK. We validated potential chromosomal anomalies using custom nanostring probes and quantitative PCR. We estimate chromosomal alterations in the schizophrenia population to be 0.42%, which is not significantly different from controls (0.26%). We identified and validated a set of four extremely large (>10 Mb) chromosomal anomalies in subjects with schizophrenia, including a chromosome 8 trisomy and deletion of the q arm of chromosome 7. These data demonstrate that chromosomal anomalies are present at low frequency in blood cells of both control and schizophrenia subjects.
Similar content being viewed by others


Introduction
Schizophrenia (SCZ) is a highly heritable, debilitating psychiatric disorder characterized by psychosis and cognitive deficits, and has a lifetime prevalence of ∼0.5–0.7%.1, 2 Recent studies into the genetic architecture of this disease have implicated both common single-nucleotide polymorphisms (SNPs) and rare copy number variants (CNVs).3, 4, 5 In particular, recent studies of rare CNVs in SCZ have identified both individuals with single events of high penetrance and abundant events across the genome. Specifically, CNVs >500 kb at 22q11–13, 15q11, 15q13, 3q29, 7q36.3 and 1q21 have been linked to SCZ in ∼1% of cases.3, 6, 7, 8, 9 In addition, it has been shown that individuals with SCZ are more likely to carry large, rare CNVs than individuals without the disorder. This ‘burden’ has also been shown to be significant in singleton CNVs – those CNVs observed only once in a particular sample.
Recent reports suggest that somatic structural changes in the nuclear genome are not uncommon and can be identified using SNP genotyping arrays,10, 11, 12 at least when using DNA from white blood cells. These studies have detected a mosaicism rate from 0.23 to 2% and mosaicism has been observed in 7 to 95% of lymphocytes based on probe intensity measures. No disease study has yet revealed a difference between cases and controls on this measure.
The purpose of this study was to determine differences between SCZ cases and controls with respect to structural changes in the genome of white blood cells, and how frequently these anomalies arise in the SCZ population, within the limitation of the detection resolution of SNP genotyping arrays. Our data suggest that both control and SCZ populations carry large chromosomal anomalies in blood cells at frequencies under 0.5%.
Materials and methods
Samples
Samples used in this study have been described previously and quality control measures have been thoroughly detailed.3, 13 Subjects in this study passed all QC measures for chip quality as with our previous analysis.3 Individuals were placed into nine groups based on site, collection date and/or Affymetrix array used (Table 1). All DNA used in the original International Schizophrenia Consortium (ISC) study or for validation in this study was derived from whole blood. Independently, we also used samples from the Molecular Genetics of Schizophrenia Genetic Association Information Network (GAIN) as an external validation set, with the caveat that increased rates of abnormality might be observed because these DNA samples were derived from Epstein–Barr virus-transformed lymphocytes. Samples were genotyped by the Genetic Analysis Platform at The Broad Institute of Harvard and MIT according to standard protocols as described previously.3 Both the Affymetrix 5.0 and 6.0 arrays were used (Table 1) and CNVs were identified using Birdseye,14 which identifies rare CNVs by integrating intensity data from neighboring probes using a hidden Markov model (HMM) on a per-individual basis. Performance is dependent on a number of factors including SNP and copy number probe density, and mean intraindividual probe variance. Covariate analysis was carried out to control for 96-well plate effects. For each copy number change, an LOD score was generated that describes the likelihood of the copy change relative to no copy change over the given interval. Large copy number changes were assessed after removing CNVs <20 kb or with LOD scores <10 and after joining segments appearing to be incorrectly split by the HMM. Rare CNVs were defined as being seen in <1% of the sample following the same procedure described previously3 in PLINK.15
We were particularly interested in subjects with a large extent of CNVs and/or those subjects with many CNVs, as both of these situations could reveal subjects with large CNVs (ie, chromosomal CNVs, or very large duplications/deletions). ‘Large extent of CNV’ refers to the situation where a single large CNV is identified, while ‘many CNVs’ refer to a situation where a single subject has an abnormally high level of CNVs, far from the group mean.
Nanostring analysis
One to four probes were designed to regions of interest by Nanostring Technologies (http://www.nanostring.com/). An oligonucleotide reporter probe was synthesized to each region ligated to one of six fluorophores ligated. A separate capture probe, which pairs with the reporter probe:DNA hybrid, then allows the complex to attach to a matrix for imaging. No amplification steps are required for this reaction. After hybridization, samples were transferred to the nCounter Prep Station where excess probes were removed and probe/target complexes were aligned and immobilized in the nCounter Cartridge. Cartridges were then placed in the nCounterDigital Analyzer for imaging. To control for the amount of DNA present across all samples, a spike-in plasmid was used and served as a positive control. Further, optimized probe pairs for 10 invariant regions of the genome were included for data normalization. Other controls were used to ensure optimal hybridization and purification efficiency.
Quantitative polymerase chain reaction
Once CNVs were identified, we randomly selected subregions from CNVs that did not overlap with subregions validated by nanostring. All primers were designed in Primer3 (http://www.primer3.com/) and all amplification products were between 100 and 180 basepairs. Using a LightCycler 480 II (Roche Diagnostics, Basel, Switzerland) and SYBR green (Promega, Madison, WI, USA), we assessed fluorescent signal over cycle time. At least two replicates were used per region and a non-varying genomic region was assessed to control for DNA concentration. All melting curves showed single peaks, suggesting that primers bound efficiently and specifically. The second derivative of the cycle threshold (Ct) was used for data analysis on factory-loaded settings. For comparison, six control CEPH subjects from the HapMap project were assessed.
Results
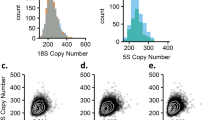
We used SNP microarray data from 3518 SCZ patients and 4238 controls3 from nine sites (Table 1) to detect potentially large chromosomal changes. Among the individuals from the complete ISC sample set,3 the median number of rare copy number changes >100 kb per individual is 1 (mean 0.9) and the median extent of CNV is 129 kb (mean 267 kb). From this set, we identified 60 outliers (defined as outside of 3 SDs from the mean extent of CNV) where the median number of rare CNVs over 100 kb per outlier individual is 3 (mean=9.7) and the median extent of CNV is 6.4 Mb (mean 17 Mb). Figures 1 and 2 show the size and distribution of rare CNVs from cases and controls in the outlier sample and Table 2 shows specific subject information.

Distribution of number of rare CNVs and total extent of rare CNV by individual. Red dots represent cases and black dots represent controls. A full color version of this figure is available at the European Journal of Human Genetics journal online.

Size and number of CNVs in the outlier sample.
We defined an outlier sample set (cases and controls, n=60) from the initial ISC sample set (case and controls, n=7756). In all, 36 outliers of 3518 cases were identified, whereas only 24 outliers of 4238 controls were identified (Fisher’s exact test P=0.026; OR=1.82). To further address whether this outlier population was enriched for individuals with SCZ, we performed logistic regression on phenotype and outlier status, accounting for the nine sites, and array type (Table 1) and gender as covariates. Overall, we found a significant over-representation of SCZ cases compared with controls (P=0.019), in line with our previous findings that suggest increased CNV burden in people with SCZ.
No cell lines were used in the above analysis and all DNA processed was isolated from blood. We had access to CNV data from the Molecular Genetics of Schizophrenia GAIN, where there were 1324 controls and 1096 SCZ cases, with the caveat that most DNA was isolated from lymphoblastoid cell lines.5 In this independent sample set, we again observed enrichment in SCZ relative to control. Using our previous criteria, we identified 22 case outliers and 12 control outliers (GAIN sample only; Fisher’s exact text P=0.024).
A number of copy number changes from this first analysis represent scattered copy changes across the genome and thus are of unclear quality and biological importance, despite passing array QC parameters. To define only those subjects with potential chromosomal anomalies (GAIN subjects are excluded from this analysis because of potential confounds from cell line artifacts), we selected subjects with very high extent of CNV (ie, suggesting a single large CNV). Of 36 cases, 15 were included in the outlier pool based on the extent of CNV in the genome, whereas 11 of 24 controls did. Four schizophrenic subjects and two wild-type subjects met the criteria for both large CNVs and many CNVs. In all, 19 cases and 13 controls were included in the outlier sample for having many CNVs. Thus, 0.42% of the total sample of cases and 0.26% of controls had large structural variation as assessed by SNP array, a number too small to detect a statistically significant difference.
To give an example of the large variation observed, we present in Table 3 nine cases and six controls with CNVs >10 Mb (the minimum size of structural variation required for karyotype analysis, under good conditions), of which one case carried two CNVs of this size. Eight of these large CNVs included a putative duplication over centromere 1 or centromere 9, and thus are of unknown validity given the lack of probes in centromeric regions (consistent signal is detected on either side of the centromere). In the GAIN sample, two cases and one control had a single CNV >10 Mb.
We sought to confirm the existence of large chromosomal anomalies, and accurate calling of structural variation from arrays, by sampling DNA from some subjects. Using two different technologies, we chose two large duplications and two large deletions from three independent cases (Figure 3). First, we used a series of custom nanostring probes to assess whether an increased or decreased signal corresponded with the genomic location of the CNV call from the arrays. As these were large events, we used multiple probes targeting different regions of the CNV (Figure 3). In each case, we were able to validate the array data for the existence of the event and an estimate of size (probes were placed across chromosome but did not cover the entirety of the called event). Second, we performed quantitative PCR on each large CNV from these subjects (Table 4). Data generated from all three technologies were in agreement (Table 4).

Non-centromeric CNVs >10 Mb identified in a refined set of outlier subjects. Red squares indicate where on the chromosome the loss (del) or gain (dup) occurred. Regions of quantitative PCR (qPCR) and nanostring probe binding are marked by small squares adjacent to chromosome images. A full color version of this figure is available at the European Journal of Human Genetics journal online.
Discussion
We used SNP genotyping arrays to call copy number gains and losses in the genome, and identified a structural variant rate of approximately 0.42% in blood cells from people with SCZ. This was a large study with close to 8000 subjects, but we could not detect a significant difference in the rate of chromosomal anomalies in cases compared with controls, where the rate of structural variation in controls was 0.23%. All CNVs described here are ‘chromosomal’ in nature, ranging from 10 Mb to complete chromosomal copy number changes. Many of these changes are not viable with life and thus subjects carrying these alterations are mosaic – that is, some cells in different tissues have post-zygotic mutations not shared by all cells from the individual.
Similar studies using SNP arrays to call large structural variation identified have similar rates of aberrations. For example, in the study by Jacobs et al,12 the rate of mosaicism in 26 136 controls was identical to our study (0.23%). In contrast, mosaicism in a bladder cancer study of 1991 individuals (957 controls) was 1.7%.11 Increased rate of mosaicism is frequently observed in cancer cells, which is likely the driver of this difference. We detected almost a twofold increase in large deletion/duplication variation in the SCZ population, suggesting that with greater power, significant differences between groups might arise. Still, it seems unlikely that such large structural variation (eg, trisomy on chromosome 8) has an affect on SCZ, nor is it clear how post-zygotic changes in non-neural tissue might influence disease.
Analysis of DNA extracted from brain rather than lymphocytes could have revealed different results. It is not clear, for example, what the relationship is between lymphocyte mosaicism and neuron mosaicism. It may be that rates in (postmitotic) neurons are very low compared with lymphocytes that continuously renew, providing more opportunities for genomic alterations over time in blood cells. While brain DNA would likely be more relevant to psychiatric disease, it is also much harder to acquire, being restricted to DNA extraction from brains stored in brain banks. The benefit of using lymphocytes is the large number of subjects that can be sampled, which is crucial given the small number of mosaic events detected. Two studies have looked at CNV events using brain DNA,16, 17 the former with a sample size of 600 and the latter using 35 brains. Neither study assessed mosaicsm, and each found only single sites of potential pathology. Still, these studies document the benefits of studying CNV events using DNA from human brain.
In this study, a majority of subjects met the criteria because of many CNVs, rather than a single large CNV. While this was a function of the filtering paradigm to find large structural variations and these subjects were removed from further analysis in this study – they did not have large structural variation – they may reflect legitimate findings and not technical artifacts. This follows with our previous finding of increased burden of CNVs in the genome; however, the sheer number of scattered CNVs across the genome of some individuals is suspiciously high, but may warrant further analysis.
Mosaicism rates are likely higher for both cases and controls than that reported here. First, we only used stringent array filtering criteria, which revealed only subjects with a large majority of cells carrying the same genotype. Specifically, mosaicism can be described in terms of percent mosaicism, that is, what proportion of cells from a given tissue are mosaic. In the quantitative PCR and nanostring validation component of our study, we observed ratios of approximately 3:2 for duplications and 2:1 for deletions, strongly suggesting that all lymphocytes carry the mutation. Thus, any subjects mosaic for, for example, 50% of cells would not have been detected. Second, our study design could only detect specific types of chromosomal alterations; we were unable to detect balanced chromosomal translocations, for example, suggesting that the true rate of chromosomal aberrations is in actuality much higher.
This study suggests that the frequency of mosaic structural variation in blood cells from people with SCZ is ∼0.42%, almost double that observed in control subjects. Still, overall numbers were too small to detect a difference between cases and controls. Future studies might assess multiple tissues for mosaicism from people with SCZ, use different technology to assess copy changes across tissues, increase sample sizes and better study those subjects with multiple small CNVs throughout their genomes.
References
Kendler KS, Gallagher TJ, Abelson JM, Kessler RC : Lifetime prevalence, demographic risk factors, and diagnostic validity of nonaffective psychosis as assessed in a US community sample. The National Comorbidity Survey. Arch Gen Psychiatry 1996; 53: 1022–1031.
Cannon TD, Kaprio J, Lonnqvist J, Huttunen M, Koskenvuo M : The genetic epidemiology of schizophrenia in a Finnish twin cohort. A population-based modeling study. Arch Gen Psychiatry 1998; 55: 67–74.
International Schizophrenia Consortium: Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455: 237–241.
Purcell SM, Wray NR, Stone JL et al: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Shi J, Levinson DF, Duan J et al: Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460: 753–757.
Mulle JG, Dodd AF, McGrath JA et al: Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet 2010; 87: 229–236.
Vacic V, McCarthy S, Malhotra D et al: Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 2011; 471: 499–503.
Brunet A, Armengol L, Heine D et al: BAC array CGH in patients with velocardiofacial syndrome-like features reveals genomic aberrations on chromosome region 1q21.1. BMC Med Genet 2009; 10: 144.
Bassett AS, Scherer SW, Brzustowicz LM : Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am J Psychiatry 2010; 167: 899–914.
Forsberg LA, Rasi C, Razzaghian HR et al: Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet 2012; 90: 217–228.
Rodriguez-Santiago B, Malats N, Rothman N et al: Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet 2010; 87: 129–138.
Jacobs KB, Yeager M, Zhou W et al: Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 2012; 44: 651–658.
Bergen SE, O’Dushlaine CT, Ripke S et al: Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol Psychiatry 2012; 17: 880–886.
Korn JM, Kuruvilla FG, McCarroll SA et al: Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 2008; 40: 1253–1260.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Ye T, Lipska BK, Tao R et al: Analysis of copy number variations in brain DNA from patients with schizophrenia and other psychiatric disorders. Biol Psychiatry 2012; 72: 651–654.
Wilson GM, Flibotte S, Chopra V, Melnyk BL, Honer WG, Holt RA : DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet 2006; 15: 743–749.
Acknowledgements
We thank all subjects for contributing DNA to our research centers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ruderfer, D., Chambert, K., Moran, J. et al. Mosaic copy number variation in schizophrenia. Eur J Hum Genet 21, 1007–1011 (2013). https://doi.org/10.1038/ejhg.2012.287
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.287
Keywords
This article is cited by
-
ARFID Genes and Environment (ARFID-GEN): study protocol
BMC Psychiatry (2023)
-
Somatic mosaicism and neurodevelopmental disease
Nature Neuroscience (2018)
-
Genome-wide association study of copy number variation with lung function identifies a novel signal of association near BANP for forced vital capacity
BMC Genetics (2016)
-
Copy number variation in schizophrenia in Sweden
Molecular Psychiatry (2014)
