
Abstract
Heterozygous mutations in dynamin 2 (DNM2) have been linked to dominant Charcot-Marie-Tooth neuropathy and centronuclear myopathy. We report the first homozygous mutation in the DNM2 protein p.Phe379Val, in three consanguineous patients with a lethal congenital syndrome associating akinesia, joint contractures, hypotonia, skeletal abnormalities, and brain and retinal hemorrhages. In vitro membrane tubulation, trafficking and GTPase assays are consistent with an impact of the DNM2p.Phe379Val mutation on endocytosis. Although DNM2 has been previously implicated in axonal and muscle maintenance, the clinical manifestation in our patients taken together with our expression analysis profile during mouse embryogenesis and knockdown approaches in zebrafish resulting in defects in muscle organization and angiogenesis support a pleiotropic role for DNM2 during fetal development in vertebrates and humans.
Similar content being viewed by others

Introduction
Developmental defects and perinatal lethal syndromes are heterogeneous diseases caused by environmental and/or genetic factors. Among these, fetal hypokinesia of genetic origin is characterized by decreased fetal movements often accompanied by muscle and lung hypoplasia and congenital contractures. Fetal hypokinesia sequence can stem from neuromuscular dysfunction and several genes implicated in postnatal neuromuscular diseases have also been associated with these developmental defects.1, 2, 3 We report the characterization of a congenital syndrome, associating fetal hypokinesia with thin bones and hemorrhages, due to a homozygous mutation in the large GTPase dynamin 2 (DNM2). Two autosomal dominant disorders have been associated with heterozygous mutations within the DNM2 gene; autosomal dominant centronuclear myopathy (ADCNM) and dominant intermediate or type 2M Charcot-Marie-Tooth neuropathies (CMT-DIB).4, 5, 6 Functional analyses support the pathological impact of the novel p.Phe379Val mutation, and downregulation experiments in zebrafish highlight a pleiotropic role for DNM2 during vertebrate development. These findings identify this congenital syndrome as allelic to dominant ADCNM and CMT-DIB.
Materials and methods
Genetic analysis
In this study, we describe a consanguineous family of Pakistani origin (second cousins) with three affected and two unaffected children. For linkage analysis, DNA samples from AFB13, AFB12, AFB2, AFB3 and AFB4 were analyzed using the GeneChip Human Mapping 250 K SNP array (Affymetrix, Santa Clara, CA, USA). Samples were prepared following the manufacturer’s instructions. Genotypes were specified by the Genotyping Console Software (GTYPE v4.0, Affymetrix) and were displayed by the HomoSNP linkage program.7
Additional information regarding methods used for histological analyses, cell culture and endocytosis assays, imaging, in vitro GTPase and membrane tubulation assays, and characterization of zebrafish models can be found in the Methods section of the Supplementary Information).
Results
Clinical and physiological description of a novel perinatal syndrome
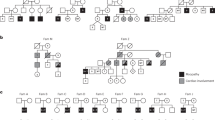
The three affected infants (AFB2, AFB3 and AFB4) were children of a consanguineous family (Figure 1a). Analysis of the patients was performed with consent from the family. They exhibited decreased fetal movements, polyhydramnios and were delivered by emergency caesarian sections with a birth weight lower than normal (Table 1 and physiological investigation in the Supplementary Information). The three patients displayed no reflexes and suffered from severe hypotonia and respiratory insufficiency from birth, while they exhibited contractures of several joints and skeletal abnormalities with thin ribs and bones (Figures 1c and e and Supplementary Figure 1). The AFB2 muscle biopsy showed small rounded fibers with some centralized nuclei, suggestive of a congenital myopathy component whereas the AFB4 biopsy showed atrophic fibers without obvious centralization of nuclei (Figure 1d). In addition, they showed retinal hemorrhages, whereas MRI analysis revealed symmetric subdural hematomas for AFB3 and blood in the subarachnoid cavity for AFB4 (Figure 1f and Table 1). AFB3 and AFB4 had pulmonary hypertension and a patent ductus arteriosus; whether these features were primary or secondary to respiratory insufficiency could not be determined. Death occurred at 5 and at 19 days of age for AFB3 and AFB4, respectively, due to progressive deterioration, whereas AFB2 died at 4 months of age due to an upper respiratory tract infection that triggered rapid deterioration. EMG analysis was suggestive of a myopathy or lower motor neuron disease for AFB2, and revealed low and extremely low conduction velocities for AFB3 and AFB4, respectively, and low CMAP’s for both AFB3 and AFB4 suggestive of hypomyelinating neuropathy or anterior horn disease (Table 1 and physiological investigation in the Supplementary Information).8

Identification of a homozygous DNM2 mutation. (a) Pedigree of the consanguineous Pakistani family investigated. Heterozygous carriers are indicated by dots. (b) Direct sequencing analysis. A single base substitution (c.1135 T>G; Genbank NM_001005360) in exon 9 of DNM2 was identified (indicated by black box) and was found homozygous in all affected and heterozygous in unaffected family members. (c) Hands of AFB3 and AFB2 display contractures. (d) Quadriceps biopsies depicting the presence of centralized nuclei in AFB2. Some fibers with centralized nuclei are indicated by arrows. Scale bars represent 50 μm. (e) Anteroposterior radiograph of the thorax and abdomen of AFB3 depicting thin ribs with normal length and bone density. Note the deep position of the endotracheal tube. (f) MRI analysis of patient AFB3. From left to right: axial T1-weighted, susceptibility, T2-weighted images of the cerebrum at the level of the basal ganglia, and axial T2-weighted image at the level of the brainstem. Bilateral hyperintense signal in the occipital subdural space (arrows) and posterior horns of the ventricles (arrowheads) with low signal intensity on the susceptibility image and hypointense signal on T2-weighted image are consistent with bilateral subdural hematomas and intraventricular hemorrhage. Symmetric areas of low signal intensity within the thalamus and globus pallidus (asterisk) are suggestive of early subacute 3–7 day-old hemorrhage. The axial T2-weighted image at the level of the brainstem indicates hemorrhagic changes in the brainstem.
Identification of a homozygous missense mutation in DNM2
All affected children had a normal karyotype. DNA analysis excluded mutations in the DMPK, SMN, MTM1 and BIN1 genes. Homozygous regions by descent were mapped, and the largest detected homozygous region common for the three affected children was a 4-Mb region on chromosome band 19p13.2 (Supplementary Figure 2 and Supplementary Table 1). Within the 141 genes mapped in this region, the DNM2 gene was selected as a candidate based on the congenital myopathy component in patient AFB2 and the suggestion of peripheral nerve involvement in AFB3 and AFB4, as distinct DNM2 heterozygous mutations were previously linked to autosomal dominant CMT peripheral neuropathy and to ADCNM.4, 6 Direct sequencing revealed a single point mutation (c.1135 T>G Genbank NM_001005360) in exon 9 at the homozygous state in all affected children (Figure 1b). Parents and unaffected children were all heterozygous for the mutation. High-resolution melting curve analysis or direct sequencing of 300 DNA samples, including 100 Pakistani controls, did not show this variation and this change is also absent from SNP databases. This mutation substitutes a conserved phenylalanine residue with valine at position 379 (p.Phe379Val) within the middle domain of DNM2 (Figure 2a).

Effect of the p.Phe379Val mutation on DNM2 function. (a) DNM2 protein domains with the p.Phe379Val point mutation indicated (top); GED, GTPase effector domain; MID, middle domain; PH, Pleckstrin homology domain; PRD, proline-rich domain. Protein alignment of the dynamin family members showing conservation of amino-acid F379 in vertebrates (bottom); ce, Caenorhabditis elegans; dm, Drosophila melanogaster; dr, Danio rerio; hs, Homo sapiens; mm, Mus musculus. (b) DNM2 protein levels are comparable between AFB2 (p.Phe379Val) and control fibroblasts as compared by western blot analysis using the in-house polyclonal antibody R2680 against DNM2. (c) p.Phe379Val patient fibroblasts exhibit reduced transferrin uptake compared with untreated controls. Arbitary units describe mean fluorescence of transferrin uptake. Error bars represent SD (P-values *0.00616 and **0.0003522). (d) The p.Phe379Val protein exhibits ∼20% lower GTPase activity using a malachite green-based assay (P-value *0.05 for three independent experiments using the Student’s t-test). Error bars represent SD. (e) Electron micrographs depicting wild-type (WT) and p.Phe379Val DNM2 incubated in the presence of small unilammellar vesicles. The p.Phe379Val protein does not tubulate liposomes and typical striated polymers of DNM2 observed in the case of WT DNM2 are absent from vesicles incubated with the DNM2p.Phe379Val protein.
Impact of the mutation at the heterozygous state
We investigated whether the heterozygous carriers showed signs of CNM or CMT. Unaffected heterozygous siblings had normal motor development and muscle strength, no contractures, and are now aged 11 and 5 (Supplementary Table 2). The mother displayed normal conduction velocities, however, heterozygous parents had impaired reflexes and analysis of the mother’s muscle biopsy revealed fiber size variation and centralized nuclei, characteristics of ADCNM whereas DNM2 localization in the mother’s biopsy appeared normal (Supplementary Table 2 and Supplementary Figure 3). This suggests that the presence of the p.Phe379Val mutation at the heterozygous state leads to mild adult onset ADCNM as seen in the mother. DNM2 levels were normal in fibroblasts from the homozygous AFB2 patient, indicative of the p.Phe379Val mutation not altering protein stability (Figure 2b).
Impact of the p.Phe379Val mutation on DNM2 function
We addressed an impact on clathrin-mediated endocytosis by measuring transferrin uptake, a process requiring dynamin action.9, 10, 11 We observed an ∼20% reduction in patient fibroblasts with the p.Phe379Val mutation (Figure 2c).10, 11 As dynamin is a GTPase that can tubulate and sever membranes,12, 13 we measured the basal GTPase activity of wild-type and p.Phe379Val DNM2 recombinant proteins and found that the p.Phe379Val mutant’s activity was reduced by 20% (Figure 2d). This mutant also failed to tubulate small unilammellar vesiscles in an in vitro assay (Figure 2e). However, the p.Phe379Val mutation had no impact on other functions of DNM2 such as centrosome cohesion and Golgi maintenance (Supplementary Figure 4).14, 15 Taken together, our observations suggest that the p.Phe379Val mutation is a hypomorphic mutation impacting on dynamin’s role in endocytosis.
DNM2 has a broad and conserved role during embryonic development
To gain more insight into the potential role of DNM2 during embryonic development, we performed in situ hybridization on mouse embryo sagital sections using a Dnm2 mRNA probe (obtained from Eurexpress).16 Dnm2 was expressed in most tissues, including the peripheral nervous system (Figure 3a, right panels), although no expression was evident in skeletal muscle or heart (for tissue profiles see Supplementary Table 3). Injection of one cell stage zebrafish embryos with a translational blocking morpholino (MO) targeting dnm2 transcripts resulted in pleiotropic phenotypes in dnm2 morphants: lethality was observed in 10% and bent-tail in 20% after 48 h post-fertilization (hpf; Supplementary Figure 5). Use of second harmonic generation (SHG) to visualize the periodic organization of myosin in sarcomeric thick filaments revealed a mild misalignment of muscle fibers in the dnm2 MO-injected embryos when compared with controls (Figure 3b, top panels), suggesting that muscle formation is affected in the dnm2 morphants. The lack of gross defects was expected as only embryos with overall unaffected morphology were analyzed to rule-out nonspecific effects due to morpholino toxicity. Moreover, analysis of the Tg(flk1:eGFP) zebrafish expressing the GFP specifically in the endothelium highlighted defects in the vascular system of embryos: the inter-segmental vessels displayed aberrant sprouting and excessive branching (Figure 3b, bottom panels). However, no alteration of the muscular innervation from the spinal cord was observed in the dnm2-MO-injected embryos, suggesting that axonal formation is not affected in the morphants (Supplementary Figure 6). These observations highlight the pleiotropic role for DNM2 during fetal development.

DNM2 expression and function during embryonic development. (a) Dnm2 mRNA can be detected in several tissues during murine embryonic development. In situ hybridization experiments were performed with a Dnm2 mRNA probe (Dnm2) on sagital sections of 14.5-day (top) and 16.5-day (bottom) mouse embryos. Controls represent embryo sections incubated with all reagents but lacking mRNA probe to measure levels of nonspecific reaction (left panels). No Dnm2 mRNA was evident in muscle at this stage. (b) Morphology of muscle fibers and vessels in the tail of control (left panels) and dnm2 MO-injected (right panels) zebrafish embryos at 72 hpf. SHG revealing the myosin fibers organization and the periodic structure of sarcomeric thick filaments (top panels). Note the mild fiber misalignment in the MO-injected embryos (arrow). Confocal reconstructions of the GFP-positive endothelial cells revealing the vascular system of the transgenic Tg(flk1:eGFP) zebrafish (middle panels). In dnm2 MO-injected embryos, the inter-segmental vessels display deficient or abnormal branching (asterisk and white arrows, respectively).
Discussion
This is the first report of a homozygous mutation in a dynamin family member. To date, only heterozygous DNM2 mutations that cause relatively mild pathologies and display tissue specificity have been identified.5, 6 The p.Phe379Val change is very likely to be the disease-causing mutation as DNM2 is located in the best linked region, the mutation segregates with the disease, biochemical and in vitro tests demonstrate an impact of this change on different aspects of DNM2 function, patient fibroblasts depict an alteration in DNM2-dependent transferrin endocytosis and an adult heterozygous carrier shows signs of CNM. DNM2 deletion in mice causes embryonic lethality (8–12 days post coitum), suggesting that DNM2 is essential for embryonic development.17 This is probably linked to the broad cellular function of DNM2 and the fact that DNM2 is ubiquitously expressed in contrast to dynamins 1 and 3.18 In addition, the perinatal syndrome described in our patients, our in situ Dnm2 hybridization observations and knockdown experiments in zebrafish are consistent with Dnm2 having a role during embryonic development.
Although features of the CNM pathology can be reproduced both by knock-in and Adeno-associated virus-mediated exogenous expression approaches for the most prevalent ADCNM-causing DNM2 mutation (R465W),19, 20 Durieux et al20 reported that the homozygous knock-in mice died shortly after birth. These observations highlight the difference in the severity of the pathology at the heterozygous and homozygous state and are consistent with the differences in the phenotypic spectrum observed for the p.Phe379Val heterozygous and homozygous carriers. The broader spectrum of clinical features seen in the p.Phe379Val homozygous patients is indicative that tissues other than skeletal muscle and the peripheral nervous system can be affected by changes in DNM2. The fact that heterozygous carriers of the p.Phe379Val mutation display a much milder phenotype, as seen for AGJ82 and AGJ83, compared with the perinatal syndrome seen in homozygous patients suggests that this mutation is leading to a partial loss-of-function. Our study along with several other reports emphasizes that DNM2 has a key role in endocytosis, and that disease-causing mutations impact on this function.9, 21, 22, 23 We speculate that alterations in endocytosis during development could explain the pleiotropic phenotype observed in patients with this congenital syndrome.
The mode of inheritance, antenatal involvement, severity of symptoms and associated skeletal abnormalities and hemorrhages stand out from features described in dominant CMT neuropathy or DCNM, suggesting that different alterations in DNM2 result in distinct phenotypes. This work also highlights the importance of DNM2 functions in various tissues during fetal development of vertebrates and humans.
References
Michalk A, Stricker S, Becker J et al: Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet 2008; 82: 464–476.
Ravenscroft G, Sollis E, Charles AK, North KN, Baynam G, Laing NG : Fetal akinesia: review of the genetics of the neuromuscular causes. J Med Genet 2011; 48: 793–801.
Sewry CA, Jimenez-Mallebrera C, Muntoni F : Congenital myopathies. Curr Opin Neurol 2008; 21: 569–575.
Bitoun M, Maugenre S, Jeannet PY et al: Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet 2005; 37: 1207–1209.
Bohm J, Biancalana V, Dechene ET et al: Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat 2012; 33: 949–959.
Zuchner S, Noureddine M, Kennerson M et al: Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet 2005; 37: 289–294.
Stoetzel C, Muller J, Laurier V et al: Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 2007; 80: 1–11.
Pareyson D, Marchesi C : Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol 2009; 8: 654–667.
Bitoun M, Durieux AC, Prudhon B et al: Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum Mutat 2009; 30: 1419–1427.
Damke H, Baba T, Warnock DE, Schmid SL : Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol 1994; 127: 915–934.
van der Bliek AM, Meyerowitz EM : Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 1991; 351: 411–414.
Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F : G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 2010; 465: 435–440.
Praefcke GJ, McMahon HT : The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 2004; 5: 133–147.
Cao H, Thompson HM, Krueger EW, McNiven MA : Disruption of Golgi structure and function in mammalian cells expressing a mutant dynamin. J Cell Sci 2000; 113 (Part 11): 1993–2002.
Thompson HM, Cao H, Chen J, Euteneuer U, McNiven MA : Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat Cell Biol 2004; 6: 335–342.
Schaeren-Wiemers N, Gerfin-Moser A : A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 1993; 100: 431–440.
Ferguson SM, Raimondi A, Paradise S et al: Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell 2009; 17: 811–822.
Ferguson SM, De Camilli P : Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 2012; 13: 75–88.
Cowling BS, Toussaint A, Amoasii L et al: Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am J Pathol 2011; 178: 2224–2235.
Durieux AC, Vignaud A, Prudhon B et al: A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet 2010; 19: 4820–4836.
Koutsopoulos OS, Koch C, Tosch V, Bohm J, North KN, Laporte J : Mild functional differences of dynamin 2 mutations associated to centronuclear myopathy and Charcot-Marie Tooth peripheral neuropathy. PLoS One 2011; 6: e27498.
Liu YW, Lukiyanchuk V, Schmid SL : Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic 2011; 12: 1620–1633.
Sidiropoulos PN, Miehe M, Bock T et al: Dynamin 2 mutations in Charcot-Marie-Tooth neuropathy highlight the importance of clathrin-mediated endocytosis in myelination. Brain 2012; 135: 1395–1411.
Acknowledgements
We thank Pietro De Camilli, Joelle Morvan, Nicolas Vitale, Dominique Massotte, Valérie Biancalana and Michel Koenig for reagents and discussion, Fabrice Klein Marc Koch, Olivia Wendling, Frederic Humbert, Emilie Peter and Isabelle Kolb-Cheynel for technical assistance, Frederic Plewniak for the homoSNP linkage program and Sjoerd van Duinen for histology images. Some control cell lines were established by the Généthon DNA and Cell Bank (Evry, France). This study was supported by grants from the Institute National de la Santé et de la Recherche Médicale, the Center National de la Recherche Scientifique, University of Strasbourg, Collège de France, the Association Française contre les Myopathies, Fondation Recherche Médicale, Agence Nationale de la Recherche and the E-rare program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Koutsopoulos, O., Kretz, C., Weller, C. et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet 21, 637–642 (2013). https://doi.org/10.1038/ejhg.2012.226
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.226
Keywords
This article is cited by
-
Differential impact of ubiquitous and muscle dynamin 2 isoforms in muscle physiology and centronuclear myopathy
Nature Communications (2022)
-
A meta-analysis of prognostic biomarkers in neonatal retinal hemorrhage
International Ophthalmology (2022)
-
Mutational and clinical spectrum of centronuclear myopathy in 9 cases and a literature review of Chinese patients
Neurological Sciences (2022)
-
A review of Dynamin 2 involvement in cancers highlights a promising therapeutic target
Journal of Experimental & Clinical Cancer Research (2021)
-
A new mutation in DNM2 gene in a large Italian family
Neurological Sciences (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}