
Abstract
Inherited ichthyoses, defined as the generalized form of Mendelian disorders of cornification, are characterized by visible scaling and/or hyperkeratosis of most or all of the skin. This etiologically and phenotypically heterogenous group of conditions is caused by mutations in various different genes important for keratinocyte differentiation and epidermal barrier function. Diagnosing a specific entity is a particular challenge for the nonspecialist presented with the common clinical scaling. For the clinician, this review outlines an algorithmic approach for utilizing diagnostic clues to narrow down the differential diagnosis and to guide further testing and treatment options.
Similar content being viewed by others
In Brief
-
Mendelian disorders of cornification (MeDOC) subtypes are distinguished clinically by the quality and distribution of scaling/hyperkeratosis, by other dermatologic and extracutaneous involvement (syndromic vs non-syndromic), by onset and by inheritance.
-
The current classification of MeDOC entities is clinically based, with the mutated gene referenced in parenthesis, for example, ARCI (TGM1) for autosomal recessive congenital ichthyosis due to transglutaminase-1 mutations.
-
Although mutations in several different genes coding for epidermal proteins have been described, they converge in a limited spectrum of MeDOC phenotypes that is invariably accompanied by skin barrier impairment driving pathogenesis.
-
To date, therapy of MeDOC is symptomatic rather than mechanism or pathogenesis based. As no cure exists for MeDOC, a major need is to develop pathogenesis-driven therapy that takes into account the skin barrier defect.
Introduction
The terms ichthyoses/Mendelian disorders of cornification (MeDOC) refer to conditions with visible scaling/hyperkeratosis of the skin. MeDOC patients generally have a high disease burden.1 In addition to clinical heterogeneity, the inborn types of MeDOC have a diverse genetic background, including autosomal dominant, autosomal recessive and X-linked inheritance (Table 1).2, 3, 4 Distinguishing between specific subtypes requires careful assessment of clinical features. Associated cutaneous and extracutaneous features, as well as disease onset and clinical course, provide important diagnostic clues. In all forms of MeDOC, the clinical presentation may be variable in both severity and in the expression of associated features, and at times additional laboratory testing including microscopy and genetic analyses is required. This review summarizes how the clinician can utilize diagnostic clues to narrow down the differential diagnosis, select additional laboratory testing and choose therapy.
Epidermal homeostasis and pathogenesis of MeDOC
An intact protective skin barrier is the ultimate function of the epidermis. The main cell type within the epidermis, the keratinocyte, exits the cell cycle and differentiates while moving from the basal layer toward the skin surface. Eventually, the outermost corneocytes, which have lost their nucleus, flake off the skin surface in a process of cell junction dissolution by proteases called desquamation.5 Essential for the epidermal barrier, corneocytes are surrounded by a lipid-enriched extracellular matrix, primarily consisting of cholesterol, free fatty acids and ceramides.6, 7, 8 The lipids are delivered to the extracellular space by specialized keratinocyte organelles, the lamellar bodies (LBs), and through a series of enzymatic and nonenzymatic steps are organized into water-impermeable extracellular lipid lamellar structures.6, 9, 10, 11 Corneocytes and lipids form the epidermal barrier that prevents outside-to-inside invasion of physical, chemical and microbial noxae, and in addition maintains fluid homeostasis by preventing inside-to-outside loss of body fluids.
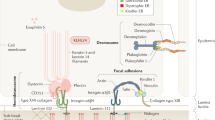
MeDOC are caused by mutations in genes for skin barrier components. These include proteins of structural importance for corneocyte formation (eg, cornified envelope proteins), cell–cell junction proteins and enzymes required for the proteolysis of cell junctions, for lipid metabolism and for DNA repair (Table 1).2, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 In the revised classification, MeDOC entities are listed using the term for the clinical entity referenced with the causative gene in parenthesis. The major clinicogenetic categories of the current MeDOC classification are summarized in Table 2 (also see Oji et al)2. However, the ultimate MeDOC classification should primarily follow ‘pathogenetic/functional’ aspects (Table 1). Mutations in different genes show phenotypic convergence in the MeDOC (Table 1), that is, different genotypes cause a relatively limited spectrum of clinical phenotypes that are neverthelesss pleiotropic (manifest as multiple phenotypic traits). This is explained by the fact that the various genetic defects found in the MeDOC all disrupt the skin barrier, eliciting a stereotypic repair mechanism.3 In MeDOC, the superimposed features of disturbed epidermal homeostasis, impaired epidermal barrier function and the ensuing repair response form the basis of the clinical phenotype (Figure 1). Notably, the barrier restoration efforts are generally unsuccessful, because the genetic disturbance remains uncorrected. Consequently, an understanding of epidermal barrier function will help the clinician to recognize the clinical criteria and subtleties that are characteristic for the individual MeDOC entities.

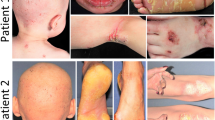
Variety of phenotypical and histological features and other dermatologic findings in different MeDOC. (a) Fine scaling phenotype in IV (FLG). (b) Moderate scaling phenotype in recessive X-linked ichthyosis (STS). (c) Large, brown scaling phenotype in ARCI (TGM1). (d) Hyperkeratosis in KI (KRT10). (e) Palmar keratoderma in LK (LOR). (f) Palmar hyperlinearity in IV (FLG). (g) Generalized fine scaling phenotype with concomitant severe erythema in NS (SPINK5). (h) Onychoschisis of fingernails in TTD, (for mutations see Table 1). (i) Tiger-tail pattern of hair under polarizing microscopy in TTD. (j) Bamboo hair (trichorrhexis invaginata) under electron microscopy in NS. (k) Collodion membrane in ARCI (TGM1). (l) Immunohistochemistry. Normal FLG staining in the granulous layer of a healthy control person (FLG +/+) and (m) absence of FLG staining in the granulous layer of a homozygous IV subject (FLG −/−). (n) Normal lamellar bilayers (arrows), corneodesmosomes (double-arrows) and corneodesmosome-derived lacunae (asterisks) in a FLG +/+ control (upper part). Foci of nonlamellar, electron-dense material (asterisks) but normal appearing corneodesmosomes (double-arrows) in a FLG −/− subject (lower part). Ruthenium tetroxide postfixation. Scale bars 0.25 μm. (o) Phase microscopy of corneocytes. Whereas corneocytes of controls appear normal after pretreatment in the ‘SDS heating test’ (see text), (p) in IV subjects a subgroup of corneocytes displays ragged fragile outlines and nuclear remnants. (q) In patients with ARCI (TGM1), corneocytes are completely destructed by detergent/heat treatment as a consequence of instability of cornified envelopes due to lacking of TGM1.
Diagnostic approach
The following clinical criteria should be used for the differential diagnosis of MeDOC: (1) the type of scaling and hyperkeratosis (Figures 1 and 2); (2) the onset and evolution of skin changes over time; (3) other dermatologic features (Figure 3); and the involvement of other organ systems (Figure 4); (4) the family history pointing toward a particular mode of inheritance or a specific genetic cause.

Scaling- and hyperkeratosis phenotype characteristics in selected examples of ichthyosis/MeDOC; for further phenotype details c.f. Oji et al.2 present  ; variable
; variable  ; not present
; not present  .
.

Other dermatologic features in selected examples of ichthyosis/MeDOC; for further phenotype details c.f. Oji et al.2 present  ; variable
; variable  ; not present
; not present  .
.

Organ involvement in selected examples of ichthyosis/MeDOC; for further phenotype details c.f. Oji et al.2 present  ; variable
; variable  ; not present
; not present  .
.
Quality and distribution of scaling and/or hyperkeratosis
Scaling and hyperkeratosis refer to thickening of the stratum corneum. Pathological scaling refers to visible flakes of stratum corneum on the surface of the skin, in contrast to invisible, physiological scaling generated by normal stratum corneum desquamation. The type of scaling, that is, fine, moderate or large (Figures 1a–c, Figure 2), provides diagnostic clues to the diagnosis of MeDOC. The term hyperkeratosis is used to describe thickened stratum corneum that does not produce visible scale, because it is not accompanied by desquamation, but gives the skin surface a ‘hardened’ texture. Examples are keratinopathic ichthyosis (KI; keratin (KRT)1, KRT2 and KRT10) with white to brown, spiny hyperkeratosis (Figure 1d) and Sjögren–Larsson syndrome (SLS) (aldehyde dehydrogenase (ALDH)3A2) with cobblestone hyperkeratosis. For both scaling and hyperkeratosis, the distribution has diagnostic significance. For example, flexural sparing is usually seen in ichthyosis vulgaris (IV) (filaggrin (FLG)) (Figure 2), whereas flexures are typically involved in ARCI. If localized to the palmoplantar skin surfaces, hyperkeratosis is termed palmoplantar keratoderma (PPK, Figure 1e, Figure 2). The presence and distribution of PPK has diagnostic relevance as exemplified in Figures 3 and 5. Other phenotype features of MeDOC include lichenification, exfoliation and erosions/blistering. The different MeDOC subtypes can present in mosaic/segmental form,22, 23, 24 which is relevant for genetic counseling, because offspring of parents with segmental forms can have generalized disease.

Algorithm for the diagnosis of congenital ichthyosis/MeDOC. EM, electron microscopy; KLICK, keratosis linearis-ichthyosis congenita-keratoderma; SDS, sodium dodecyl sulfate.
Evolution of skin changes
The neonatal presentation of MeDOC at birth provides clues for narrowing down the differential diagnosis.25 Neonates can present with congenital ichthyosiform erythroderma (CIE) or with a collodion membrane (parchment-like membrane covering the skin at birth and dissolving during the first days or weeks of life (Figure 1k)). If the history is taken years later, it is often difficult to obtain a precise description of the neonatal presentation (unless the restrictive, armorlike scaling of harlequin ichthyosis (ATP-binding cassette subfamily A12 (ABCA12)) was present).26, 27 In a minority of cases (<10%), the neonatal phenotype (largely) resolves within weeks to months post birth (termed self-improving collodion ichthyosis, previously self-healing collodion baby or pleomorphic ichthyosis).28, 29 However, this occurs in a maximum of 10% of cases. The majority of the congenital phenotypes transform into generalized scaling that typically persists throughout life.30, 31 Polyhydramnion associated with prematurity and post-natal failure to thrive are clinical features relevant for the diagnosis of ichthyosis-prematurity syndrome (FATP4).32 KI (KRT1, KRT2 and KRT10; in its generalized form also called epidermolytic ichthyosis, bullous ichthyosis, bullous CIE Brocq or ichthyosis bullosa of Siemens) presents with generalized blistering at birth that later transforms into hyperkeratosis (Figure 1d). Generally, in MeDOC with a severe phenotype there is an increased risk of growth failure due to the defective skin barrier and increased energy consumption.33
In other instances, the scaling phenotype manifests with delayed onset within weeks to months after birth. The paradigmatic example of a delayed onset condition is IV (FLG) with fine white scale (Figures 1a and f, Figure 2).34 Other examples for delayed onset MeDOC include Refsum syndrome (phytanoyl-CoA hydroxylase (PHYH)/peroxisome biogenesis factor 7 (PEX7)) and cerebral-dysgenesis-neuropathy-ichthyosis-palmoplantar keratoderma (CEDNIK, synaptosomal-associated protein 29 (SNAP29)) (Figure 2).
Other dermatologic features and organ involvement
The association of scaling/hyperkeratosis with erythema is an important distinguishing feature that is seen in CIE, Netherton syndrome (NS, serine protease inhibitor, kazal-type 5 (SPINK5)) (Figure 1g, Figure 3, Figure 5) and KI (KRT1, KRT2 and KRT10). Abnormalities of the nails, hair and mucous membranes are characteristic of several MeDOC subtypes (Figure 3), for example, onychoschisis of fingernails and tiger-tail pattern of hair under polarizing microscopy in trichothiodystrophy (TTD) (for mutations see Table 1; TTD, Figures 1h and i), trichorhexis in NS (SPINK5), alopecia/atrichia and frequent mucocutaneous infections in KID syndrome (gap junction protein-beta 2(GJB2)) (Figure 2), ear deformations in ARCI (for mutations see Table 1).
MeDOC can present as a non-syndromic (manifesting only in the skin) or as a syndromic disease (with involvement of other organ systems). A prototypic non-syndromic form is KI (KRT1, KRT2 and KRT10), which does not involve any other organ apart from the skin. At the syndromic end of the spectrum, scaling is a variable clinical feature of multi-system disorders such as Refsum syndrome (PHYH/PEX7), or the very itchy SLS (ALDH3A2), which also present with central nervous system involvement.35, 36, 37 As an intermediate type, recessive X-linked ichthyosis (XLI, steroid sulfatase (STS)) can be limited to the skin but may also show extracutaneous features such as undescended testes/cryptorchidism (although this seems to occur less frequently than previously thought) and corneal opacities (Figure 4). Detailed phenotype descriptions of the various entities are available from a recent review by Oji et al.2
Laboratory testing
The phenotype and the family history guide the initial steps of the diagnostic process before additional laboratory testing is carried out.
Light microscopy
Only a subset of MeDOC entities has pathognomonic histological features such as IV (FLG) where a reduced or absent granular layer is evident. KI (KRT1, KRT2, KRT10) shows enlarged, pale keratinocytes with abundant keratohyalin granules.2, 38, 39, 40 Specific for neutral lipid storage disease with ichthyosis (ABHD5; also termed Chanarin-Dorfman syndrome) are lipid vacuoles within polymorphonuclear leukocytes and monocytes (oil red stain: Jordan’s anomaly).41 Parakeratosis, the retention of nuclei in stratum corneum cells, is observed in loricrin keratoderma (LK, loricrin (LOR) (also termed Vohwinkel syndrome with ichthyosis)) and NS (SPINK5).42, 43 Immunohistochemical staining confirms FLG deficiency in IV (Figures 1l and m)44 and reveals absent or reduced expression of lympho-epithelial kazal-type-related inhibitor and SNAP29 in NS and CEDNIK, respectively.45, 46, 47 In addition, light microscopy of hair shafts demonstrates ‘bamboo hairs’ (trichorrhexis invaginata) in NS (Figure 1j)48 and polarization microscopy of TTD patient hairs reveals a tiger-tail pattern that corresponds to the diagnostic low sulfur protein content (Figure 1i).49, 50
Electron microscopy
Electron microscopy helps to narrow down the list of differential diagnoses or even rules out certain types of MeDOC.6 A combination of osmium tetroxide and ruthenium tetroxide post-fixation enables improved visualization of postsecretory changes in LB contents and extracellular lamellar lipids.51 For example, in IV (FLG) a pronounced decrease in keratohyaline granules is associated with abnormal LB secretion and lamellar/non-lamellar phase separation (Figure 1n), presumably due to altered enzyme activities.39, 52 XLI (STS) typically shows retained corneodesmosomes and phase separation of lipids in the stratum corneum interstices.53 Harlequin ichthyosis (ABCA12) and CEDNIK (SNAP29) exhibit abnormal LBs54 due to impaired lipid transport and abnormal intercellular lamellae in the stratum corneum.55 Gaucher disease (glucocerebrosidase (GBA)) is characterized by impaired processing of the extracellular lipids derived from the LBs.56 Disruption of the KRT cytoskeleton, with detachment from desmosomal plaques, perinuclear shells, and LB secretion defects, are observed in KI.57, 58, 59, 60, 61 LK (LOR) and ARCI (TGM1) are characterized by reduced thickness of the cornified envelopes ultrastructurally.42, 43, 62, 63, 64 In contrast, aberrant vesicular structures indicate NIPA-like domain containing 4 (also known as ichthyin) mutations in ARCI.65 Furthermore, trilamellar membrane aggregations in the stratum corneum and stratum granulosum are characteristic of ichthyosis prematurity syndrome (FATP4).51, 66, 67, 68 NS displays asymmetric cleavage of corneodesmosomes and a blockage of LB secretion.69
Other laboratory tests
To screen for TGM1-deficiency in ARCI, an immunochemical transglutaminase activity assay is carried out on unfixed cryosections.70 Alternatively, superficial stratum corneum material is subjected to a ‘sodium dodecyl sulfate (SDS) heating test’, in which a lack of cross-linked cornified envelopes is substantiated by reduced integrity (Figures 1o–p)39 (after boiling minced scales in a buffer-solution consisting of 2% SDS, ethylenediaminetetraacetic acid, dithiothreitol and Tris HCl for 10 min, and centrifuging, the corneocyte suspension is assessed by light microscopy in the Nomarsky mode). Unaffected cornified envelopes maintain integrity after boiling in SDS, as compared with TGM1-deficient material (Figure 1q).70, 71 STS deficiency underlying XLI is demonstrated by reduced arylsulfatase-C activity of leukocytes or cultured fibroblasts.72, 73, 74, 75 Elevated blood cholesterol sulfate levels with increased mobility of beta-lipoproteins are also diagnostic.76 In Refsum syndrome (PHYH/PEX7), increased phytanic acid and reduced pristanic acid levels in plasma are diagnostic in conjunction with the clinical phenotype.77 Patients with Gaucher disease (GBA) show elevated chitotriosidase in serum and characteristic Gaucher cells in the bone marrow; the diagnosis is confirmed through enzyme studies (reduced GBA activity in leukocytes or cultured fibroblasts).56 Elevated levels of 8-dehydrocholesterol in the Conradi–Hünermann–Happle syndrome (EBP) are recognized through specific sterol analyses.78 Finally, reduced ALDH activity in leukocytes or cultured fibroblasts is diagnostic in SLS (ALDH 3A2).35, 77, 79, 80
Genetic analyses
Genetic analyses are used to unequivocally establish the molecular basis of the disorder once clinical features and laboratory tests have provided sufficient information to reduce the number of probable underlying genetic causes. Candidate genes and testing facilities are compiled in Oji et al2 and are available online (netzwerk-ichthyose.de/fileadmin/nirk/uploads/Molecular_analyses_for_ichthyoses_NEU_2011.pdf; geneskin.eu; orpha.net; genetests.org). In cases where known mutations are not detected, genetic linkage analysis may be considered in appropriate families. Despite advances in molecular biology, techniques required to establish the diagnosis of MeDOC based on molecular criteria are not readily available in every geographic location of the world. Therefore, clinically-based algorithms (see above and Figures 2, 3, 4, 5) remain at the core of differential diagnostic considerations and focus the clinician’s mind to make efficient use of resources. Importantly, the diagnostic process should be discussed with the patient ahead of time to set realistic expectations, psychological support and contact to patient advocacy groups should be offered (eg, ichthyose.eu, firstskinfoundation.org).
Genetic counseling
Once a MeDOC has been diagnosed, genetic counseling provides affected individuals and/or relatives with information about diagnosis, prognosis, underlying pathogenesis if known and the mode of inheritance. The recurrence risks for future siblings and other family members may be an important issue for patients and their parents. Risk assessment is based on the probable modes of inheritance or empirical studies if the underlying genetic defect is not known. Consequences and limitations of planned genetic analyses as well as the implications of normal results should be explained.
Clues to circumvent unnecessary diagnostic investigations
To facilitate and streamline the diagnostic process, we present Figures 2, 3, 4, 5 that visualize the differential diagnostic criteria. Detailed descriptions of each entity are not provided here because of space restrictions, but can be obtained from a recent consensus classification.2 For example, to distinguish ARCI (for mutations see Table 1) from XLI (STS) and IV (FLG), a history of autosomal recessive inheritance and flexural involvement points to ARCI, which should then be primarily tested. Light microscopy for hair abnormalities can in some cases focus the differential diagnosis, for example, TTD (for mutations see table 1) vs NS (SPINK5) can be distinguished (tiger tail pattern vs trichorrhexis invaginata) and NS shows distinctive ultrastructural features, eliminating the need for broader testing (see above). As shown in Figure 5, the differential diagnosis of congenital ichthyosis can be narrowed based on clinical grounds combined with microscopy, biochemical and genetic testings. To distinguish syndromic forms, neurological, ophthalmological and ear, nose and throat work up is helpful; X-rays reveal bone changes (in Conradi–Hünermann–Happle syndrome (EBP), clinical exam and ultrasound imaging reveal hepatosplenomegaly in Gaucher disease (GBA)).
Therapy and implications of barrier impairment
At present, treatment of most MeDOC is rarely type-specific or corrective.81, 82 Instead, therapy of MeDOC is symptomatic and mostly based on patient’s experiences rather than disease pathogenesis. Often patients are experts in their individual needs and the role of the physician is to provide guidance on general treatment principals and information about potential interactions and adverse effects. It is important to keep the goals realistic and focused as no curative treatment is available. These include (1) to maintain or improve function (prevent excessive transepidermal water loss, prevent contractures), (2) to ameliorate symptoms, (3) to improve appearance and comfort, as well as (4) to treat complications (eg, secondary infections).
In infants, clinicians have to consider the functional consequences of the epidermal barrier defect. Caloric insufficiency due to evaporative energy losses places infants with severe phenotypes at risk for growth failure and requires early intervention.33 Neonates with severe congenital phenotypes require intensive care using humidified incubators to avoid temperature instability and hypernatremic dehydration; vigilance for signs of cutaneous infections and septicemia is essential. According to a case series of 17 collodion babies,83 intravenous lines and early emollient use should be avoided as far as possible to prevent infections.
During the course of the disease, MeDOC therapy usually includes soaking baths to hydrate the thickened stratum corneum, keratolytics (in infants, do not use topical salicylic acid, because of a potential for systemic adverse effects), mechanical methods and emollients (Table 3).84, 85, 86, 87 Mechanical removal of thickened stratum corneum after a soaking bath using pumice stones is simple and effective. Special locations such as the outer ear canals require professional help with regular removal of scaling/hyperkeratosis. Emollients, mostly consisting of non-physiological lipid mixtures, were traditionally selected by trial and error, but recent developments open possibilities for a pathogenesis-based treatment using physiological lipids that reflect the lipid composition of the stratum corneum (mainly cholesterol, free fatty acids and sphingolipids).88, 89 Recently, specific replacement (substitution) of the defective lipid species has been described in congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome, a prototypic example of pathogenesis-based, subtype-specific therapy.90 Any MeDOC treatment requires careful tailoring to the subtype diagnosis and the severity of the disease. For example, a strong keratolytic, that is, lactic acid (3–5%), may be beneficial for subtypes of ARCI (for mutations see Table 1) with thick adherent scales, but may be detrimental for patients with KI (KRT1, KRT2 and KRT10), because hyperkeratotic lesions are often replaced by erosions.
Another mainstay of MeDOC therapy is the modulation of epidermal differentiation by nuclear hormone receptor ligands such as topical vitamin D, topical and oral retinoids. Topical retinoids are particularly helpful in certain anatomic locations including palms, soles and eyelids (eg, treatment of ectropion). Systemic retinoids, while widely used to treat severe disease, require experienced physicians and close follow up of patients.91 Before the administration of systemic retinoids, a thorough discussion of expected outcomes and possible adverse effects is required. The lowest possible dose producing desired clinical outcomes needs to be titrated, and 1 mg/kg of isotretinoin or 0.5 mg/kg of acitretin should not be exceeded. Adverse effects include mucocutanous toxicities (eg, cheilitis and eye irritation), hair loss, hyperostoses and laboratory abnormalities in blood cell counts, transaminases and serum lipids. Retinoic acid metabolism blocking agents such as liarozole, which at present have an orphan drug status in the treatment of MeDOC, have shown an effectiveness equal to oral retinoids, and a favorable tolerability profile.92, 93, 94 However, further clinical trials are needed to confirm these findings.
It has become increasingly evident that the permeability barrier – the most critical function of the stratum corneum – is impaired in most forms of MeDOC. A better understanding of the pathogenic mechanism of each disorder from the causative genetic lesion to subsequent alterations in the stratum corneum and in skin barrier function is warranted for the development of more specific and effective therapies. New therapeutic approaches that take into account the interplay between causative genetic defect and compensatory epidermal responses are likely to significantly improve the quality of life of the affected patients.95
References
Styperek AR, Rice ZP, Kamalpour L et al. Annual direct and indirect health costs of the congenital ichthyoses. Pediatr Dermatol 2010; 27: 325–336.
Oji V, Tadini G, Akiyama M et al. Revised nomenclature and classification of inherited ichthyoses. results of the First Ichthyosis Consensus Conference in Soreze 2009 J Am Acad Dermatol 2010; 63: 607–641.
Schmuth M, Gruber R, Elias PM, Williams ML : Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol 2007; 23: 231–256.
Oji V, Traupe H : Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16: 349–359.
Jonca N, Leclerc EA, Caubet C, Simon M, Guerrin M, Serre G : Corneodesmosomes and corneodesmosin: from the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur J Dermatol 2011; 21 (Suppl 2): 35–42.
Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M : Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res 2008; 49: 697–714.
Elias PM, Brown BE, Fritsch P, Goerke J, Gray GM, White RJ : Localization and composition of lipids in neonatal mouse stratum granulosum and stratum corneum. J Invest Dermatol 1979; 73: 339–348.
Madison KC : Barrier function of the skin: ‘la raison d’etre’ of the epidermis. J Invest Dermatol 2003; 121: 231–241.
Elias PM, Goerke J, Friend DS : Mammalian epidermal barrier layer lipids: composition and influence on structure. J Invest Dermatol 1977; 69: 535–546.
Elias PM, Friend DS : The permeability barrier in mammalian epidermis. J Cell Biol 1975; 65: 180–191.
Nemes Z, Steinert PM : Bricks and mortar of the epidermal barrier. Exp Mol Med 1999; 31: 5–19.
Fischer J : Autosomal recessive congenital ichthyosis. J Invest Dermatol 2009; 129: 1319–1321.
Blaydon DC, Nitoiu D, Eckl KM et al. Mutations in CSTA, encoding cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell-cell adhesion. Am J Hum Genet 2011; 89: 564–571.
Oji V, Eckl KM, Aufenvenne K et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet 2010; 87: 274–281.
Arin MJ, Oji V, Emmert S et al. Expanding the keratin mutation database: novel and recurrent mutations and genotype-phenotype correlations in 28 patients with epidermolytic ichthyosis. Br J Dermatol 2011; 164: 442–447.
Israeli S, Khamaysi Z, Fuchs-Telem D et al. A mutation in LIPN, encoding epidermal lipase N, causes a late-onset form of autosomal-recessive congenital ichthyosis. Am J Hum Genet 2011; 88: 482–487.
Huber M, Rettler I, Bernasconi K et al. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 1995; 267: 525–528.
Russell LJ, DiGiovanna JJ, Rogers GR et al. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet 1995; 9: 279–283.
Rothnagel JA, Dominey AM, Dempsey LD et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992; 257: 1128–1130.
Maestrini E, Monaco AP, McGrath JA et al. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel's syndrome. Nat Genet 1996; 13: 70–77.
Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E : The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell 1992; 70: 811–819.
Happle R : X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl 2006; 95: 16–23.
Paller AS : Expanding our concepts of mosaic disorders of skin. Arch Dermatol 2001; 137: 1236–1238.
Choate KA, Lu Y, Zhou J et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science 2010; 330: 94–97.
Vahlquist A, Ganemo A, Pigg M, Virtanen M, Westermark P : The clinical spectrum of congenital ichthyosis in Sweden: a review of 127 cases. Acta Derm Venereol Suppl (Stockh) 2003; 213: 34–47.
Kelsell DP, Norgett EE, Unsworth H et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet 2005; 76: 794–803.
Rajpopat S, Moss C, Mellerio J et al. Harlequin ichthyosis: a review of clinical and molecular findings in 45 cases. Arch Dermatol 2011; 147: 681–686.
Vahlquist A, Bygum A, Ganemo A et al. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J Invest Dermatol 2010; 130: 438–443.
Vahlquist A : Pleomorphic ichthyosis: proposed name for a heterogeneous group of congenital ichthyoses with phenotypic shifting and mild residual scaling. Acta Derm Venereol 2010; 90: 454–460.
Eckl KM, de Juanes S, Kurtenbach J et al. Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J Invest Dermatol 2009; 129: 1421–1428.
Grall A, Guaguere E, Planchais S et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet 2012; 44: 140–147.
Klar J, Schweiger M, Zimmerman R et al. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet 2009; 85: 248–253.
Moskowitz DG, Fowler AJ, Heyman MB et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr 2004; 145: 82–92.
Smith FJ, Irvine AD, Terron-Kwiatkowski A et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 2006; 38: 337–342.
Rizzo WB : Inherited disorders of fatty alcohol metabolism. Mol Genet Metab 1998; 65: 63–73.
Rizzo WB : Sjogren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab 2007; 90: 1–9.
Ganemo A, Jagell S, Vahlquist A : Sjogren-larsson syndrome: a study of clinical symptoms and dermatological treatment in 34 Swedish patients. Acta Derm Venereol 2009; 89: 68–73.
Scheimberg I, Harper JI, Malone M, Lake BD : Inherited ichthyoses: a review of the histology of the skin. Pediatr Pathol Lab Med 1996; 16: 359–378.
Gruber R, Elias PM, Crumrine D et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol 2011; 178: 2252–2263.
Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson-Bostom L : Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol 2008; 59: 86–90.
Williams ML, Koch TK, O’Donnell JJ et al. Ichthyosis and neutral lipid storage disease. Am J Med Genet 1985; 20: 711–726.
Korge BP, Ishida-Yamamoto A, Punter C et al. Loricrin mutation in Vohwinkel's keratoderma is unique to the variant with ichthyosis. J Invest Dermatol 1997; 109: 604–610.
Ishida-Yamamoto A : Loricrin keratoderma: a novel disease entity characterized by nuclear accumulation of mutant loricrin. J Dermatol Sci 2003; 31: 3–8.
Gruber R, Janecke AR, Fauth C, Utermann G, Fritsch PO, Schmuth M : Filaggrin mutations p.R501X and c.2282del4 in ichthyosis vulgaris. Eur J Hum Genet 2007; 15: 179–184.
Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 2005; 77: 242–251.
Ong C, O'Toole EA, Ghali L et al. LEKTI demonstrable by immunohistochemistry of the skin: a potential diagnostic skin test for Netherton syndrome. Br J Dermatol 2004; 151: 1253–1257.
Raghunath M, Tontsidou L, Oji V et al. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J Invest Dermatol 2004; 123: 474–483.
Netherton EW : A unique case of trichorrhexis nodosa; bamboo hairs. AMA Arch Derm 1958; 78: 483–487.
Itin PH, Sarasin A, Pittelkow MR : Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol, 2001; 44: 891–920, quiz 1–4.
Sass JO, Skladal D, Zelger B, Romani N, Utermann B : Trichothiodystrophy: quantification of cysteine in human hair and nails by application of sodium azide-dependent oxidation to cysteic acid. Arch Dermatol Res 2004; 296: 188–191.
Elias PM, Williams ML, Crumrine D, Ichthyoses Clinical Schmuth M. : Biochemical, pathogenic and diagnostic assessment. Curr Probl Dermatol 2010; 39.
Sandilands A, Terron-Kwiatkowski A, Hull PR et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 2007; 39: 650–654.
Elias PM, Crumrine D, Rassner U et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol 2004; 122: 314–319.
Elias PM, Fartasch M, Crumrine D, Behne M, Uchida Y, Holleran WM : Origin of the corneocyte lipid envelope (CLE): observations in harlequin ichthyosis and cultured human keratinocytes. J Invest Dermatol 2000; 115: 765–769.
Akiyama M, Sugiyama-Nakagiri Y, Sakai K et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 2005; 115: 1777–1784.
Holleran WM, Ziegler SG, Goker-Alpan O et al. Skin abnormalities as an early predictor of neurologic outcome in Gaucher disease. Clin Genet 2006; 69: 355–357.
Anton-Lamprecht I : Prenatal diagnosis of genetic disorders of the skin by means of electron microscopy. Hum Genet 1981; 59: 392–405.
Anton-Lamprecht I : Genetically induced abnormalities of epidermal differentiation and ultrastructure in ichthyoses and epidermolyses: pathogenesis, heterogeneity, fetal manifestation, and prenatal diagnosis. J Invest Dermatol 1983; 81: 149s–156s.
Ishida-Yamamoto A, McGrath JA, Judge MR, Leigh IM, Lane EB, Eady RA : Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). J Invest Dermatol 1992; 99: 19–26.
Muller FB, Huber M, Kinaciyan T et al. A human keratin 10 knockout causes recessive epidermolytic hyperkeratosis. Hum Mol Genet 2006; 15: 1133–1141.
Schmuth M, Yosipovitch G, Williams ML et al. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol 2001; 117: 837–847.
Schmuth M, Fluhr JW, Crumrine DC et al. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis). J Invest Dermatol 2004; 122: 909–922.
Anton-Lamprecht I : Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. J Invest Dermatol 1994; 103: 6S–12S.
Elias PM, Schmuth M, Uchida Y et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol 2002; 11: 248–256.
Dahlqvist J, Klar J, Hausser I et al. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet 2007; 44: 615–620.
Khnykin D, Ronnevig J, Johnsson M et al. Ichthyosis prematurity syndrome: Clinical evaluation of 17 families with a rare disorder of lipid metabolism. J Am Acad Dermatol 2012; 66: 606–616.
Inhoff O, Hausser I, Schneider SW et al. Ichthyosis prematurity syndrome caused by a novel fatty acid transport protein 4 gene mutation in a German infant. Arch Dermatol 2011; 147: 750–752.
Melin M, Klar J, Gedde-Dahl T et al. A founder mutation for ichthyosis prematurity syndrome restricted to 76 kb by haplotype association. J Hum Genet 2006; 51: 864–871.
Hachem JP, Wagberg F, Schmuth M et al. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol 2006; 126: 1609–1621.
Raghunath M, Hennies HC, Velten F et al. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch Dermatol Res 1998; 290: 621–627.
Jeon S, Djian P, Green H : Inability of keratinocytes lacking their specific transglutaminase to form cross-linked envelopes: absence of envelopes as a simple diagnostic test for lamellar ichthyosis. Proc Natl Acad Sci USA 1998; 95: 687–690.
Meyer JC, Weiss H, Grundmann HP, Wursch TG, Schnyder UW : Deficiency of arylsulfatase C in cultured skin fibroblasts of X-linked ichthyosis. Hum Genet 1979; 53: 115–116.
Shapiro LJ : Steroid sulfatase deficiency and X-linked ichthyosis. Clin Biochem 1979; 12: 205.
Meyer JC, Grundmann HP : Arylsulfatase C activities in skin preparations of X-linked and autosomal dominant ichthyosis. Arch Dermatol Res 1980; 269: 213–215.
Webster D, France JT, Shapiro LJ, Weiss R : X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet 1978; 1: 70–72.
Epstein EH, Krauss RM, Shackleton CH : X-linked ichthyosis: increased blood cholesterol sulfate and electrophoretic mobility of low-density lipoprotein. Science 1981; 214: 659–660.
Paller AS : Laboratory tests for ichthyosis. Dermatol Clin 1994; 12: 99–107.
Has C, Seedorf U, Kannenberg F et al. Gas chromatography-mass spectrometry and molecular genetic studies in families with the Conradi-Hunermann-Happle syndrome. J Invest Dermatol 2002; 118: 851–858.
Goldsmith LA, Baden HP, Canty TG : Sjogren-Larsson syndrome. Diversity of cutaneous manifestations. Acta Derm Venereol 1971; 51: 374–378.
Taube B, Billeaud C, Labreze C, Entressangles B, Fontan D, Taieb A : Sjogren-Larsson syndrome: early diagnosis, dietary management and biochemical studies in two cases. Dermatology 1999; 198: 340–345.
Vahlquist A, Ganemo A, Virtanen M : Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol 2008; 88: 4–14.
Fleckman P : Management of the ichthyoses. Skin Therapy Lett 2003; 8: 3–7.
Van Gysel D, Lijnen RL, Moekti SS, de Laat PC, Oranje AP : Collodion baby: a follow-up study of 17 cases. J Eur Acad Dermatol Venereol 2002; 16: 472–475.
Oji V, Traupe H : Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 2009; 10: 351–364.
Epstein E : Evaluation of sodium chloride ointments in the treatment of ichthyosis vulgaris. Arch Dermatol 1960; 82: 998–1001.
Ganemo A, Virtanen M, Vahlquist A : Improved topical treatment of lamellar ichthyosis: a double-blind study of four different cream formulations. Br J Dermatol 1999; 141: 1027–1032.
Gnemo A, Vahlquist A : Lamellar ichthyosis is markedly improved by a novel combination of emollients. Br J Dermatol 1997; 137: 1017–1018.
Williams ML, Elias PM : Enlightened therapy of the disorders of cornification. Clin Dermatol 2003; 21: 269–273.
Wang F, Man MQ, Elias PM : A lipid mixture improves skin hydration in ichthyosis vulgaris. Int J Dermatol 1997; 36: 876–877.
Paller AS, van Steensel MA, Rodriguez-Martin M et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol 2011; 131: 2242–2248.
DiGiovanna JJ, Mauro T, Milstone LM, Schmuth M, Toro J : Use of systemic retinoids for treatment of ichthyoses. J Dermatol Treat, in press.
Verfaille CJ, Borgers M, van Steensel MA : Retinoic acid metabolism blocking agents (RAMBAs): a new paradigm in the treatment of hyperkeratotic disorders. J Dtsch Dermatol Ges 2008; 6: 355–364.
Pavez Lorie E, Chamcheu JC, Vahlquist A, Torma H : Both all-trans retinoic acid and cytochrome P450 (CYP26) inhibitors affect the expression of vitamin A metabolizing enzymes and retinoid biomarkers in organotypic epidermis. Arch Dermatol Res 2009; 301: 475–485.
van Steensel MA : Emerging drugs for ichthyosis. Expert Opin Emerg Drugs 2007; 12: 647–656.
Ganemo A, Lindholm C, Lindberg M, Sjoden PO, Vahlquist A : Quality of life in adults with congenital ichthyosis. J Adv Nurs 2003; 44: 412–419.
Acknowledgements
We are grateful to our patients who not only have taught us about the objective features of their disease, but also about the subjective aspects and their coping strategies. MeDOC studies in our group cited in this review have been funded by the Medical Research Fund Tirol (MFF), the Austrian Science Fund (FWF), the German Research Fund (DFG), the European Union (COST Action) and the National Institute of Health (NIH).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Schmuth, M., Martinz, V., Janecke, A. et al. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur J Hum Genet 21, 123–133 (2013). https://doi.org/10.1038/ejhg.2012.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.121
Keywords
This article is cited by
-
Ichthyosis
Nature Reviews Disease Primers (2023)
-
Update Ichthyosen: Differenzialdiagnostik und Therapie
hautnah dermatologie (2022)
-
Update Ichthyosen
hautnah (2021)
-
Ichthyosis: case report in a Colombian man with genetic alterations in ABCA12 and HRNR genes
BMC Medical Genomics (2021)
-
Mutation in ALOX12B likely cause of POI and also ichthyosis in a large Iranian pedigree
Molecular Genetics and Genomics (2020)
