
Abstract
Speech sound disorders are heterogeneous conditions, and sporadic and familial cases have been described. However, monogenic inheritance explains only a small proportion of such disorders, in particular in cases with childhood apraxia of speech (CAS). Deletions of <5 Mb involving the 12p13.33 locus is one of the least commonly deleted subtelomeric regions. Only four patients have been reported with such a deletion diagnosed with fluorescence in situ hybridisation telomere analysis or array CGH. To further delineate this rare microdeletional syndrome, a French collaboration together with a search in the Decipher database allowed us to gather nine new patients with a 12p13.33 subtelomeric or interstitial rearrangement identified by array CGH. Speech delay was found in all patients, which could be defined as CAS when patients had been evaluated by a speech therapist (5/9 patients). Intellectual deficiency was found in 5/9 patients only, and often associated with psychiatric manifestations of various severity. Two such deletions were inherited from an apparently healthy parent, but reevaluation revealed abnormal speech production at least in childhood, suggesting variable expressivity. The ELKS/ERC1 gene, which encodes for a synaptic factor, is found in the smallest region of overlap. These results reinforce the hypothesis that deletions of the 12p13.33 locus may be responsible for variable phenotypes including CAS associated with neurobehavioural troubles and that the presence of CAS justifies a genetic work-up.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is a major social, educational and health problem affecting 3% of the population. Speech delay is frequently associated with intellectual deficiency, but is rarely the predominant symptom. Specific language impairment is the most frequently diagnosed form of developmental language disorder, affecting up to 7% of 5- and 6-year-old children.1 There is a considerable variation in the profile of the linguistic deficits observed and the functions affected, which may be expressive, receptive or both.2 Among speech sound disorders, childhood apraxia of speech (CAS) also labelled developmental verbal dyspraxia (DVD) generally refers to the productive aspects of verbal communication.3 CAS/DVD is defined by the association of: (1) inconsistent error production on both consonants and vowels across repeated productions of syllables or words; (2) lengthened and impaired coarticulatory transitions between sounds and syllables; and (3) inappropriate prosody.4 Point mutations and chromosomal abnormalities that affect FOXP2 were the first known genetic bases in such phenotypes.5, 6, 7, 8, 9, 10 The affected persons also had variable levels of impairment in expressive and receptive language, extending to abnormal production and comprehension of grammar. FOXP2 disruptions were found in approximately 2% of a recruitment sample with ‘DVD’ (1/49).6, 10, 11 Other causative genes were secondarily described, associating speech disorders to neurobehavioural abnormalities, including the CNTNAP2, FOXP1 and SRPX2 genes.12, 13, 14, 15, 16, 17
Herein, we report on a series of nine patients with 12p subtelomeric deletions, including two familial cases with severe speech sound disorders, defined as CAS/DVD when evaluated. We therefore aimed to further clinically delineate the ‘12p13.33 microdeletion syndrome’ and determined the smallest region of overlap and a candidate gene for speech sound disorders strongly suggesting CAS/DVD.
Methods
French array-CGH network
A collaborative study was set up to colligate all of the French cases carrying a 12p13.33 microdeletion. Informed consent was obtained for all tested patients. These platforms used either the Human Genome CGH Microarray 44, 105, 180 or 244K from Agilent according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA, USA). Data were processed with feature extraction (v. 9.1) software and the results were analysed with CGH analytics (v. 4.0) software (Agilent Technologies) in the Hg18 genome assembly. When a 12p13.33 deletion was identified through array CGH, the anomaly was confirmed either by fluorescence in situ hybridisation (FISH) or quantitative PCR (qPCR).18 In cases of inherited deletion, the size of the parental deletion was determined by array CGH.
European decipher database
The decipher database (http://decipher.sanger.ac.uk) was interrogated for 12p13.33 deletions and allowed us to find new cases in England (patient 6) and Canada (patient 8). An SNP array study using the Affymetrix SNP6.0 array was used and the deletion was confirmed on FISH studies using the BAC probe RP11-359B12. Parental analyses were performed using FISH analyses.
Specialised evaluation of speech
The patients from family 1 and 2 were evaluated by a specialised speech therapist from a reference centre for speech disorders. Patient 6 and 9 were evaluated by their speech therapist, using the standardised scales discussed below. In order to determine the cause of speech production troubles, specific tests were performed including comparison between receptive and expressive abilities, a search for errors on consonants and vowels in repeated production of syllables or words, and its correlation with the length of the words (according to the BALE scale, ELO battery, ERTL-4 scale),19, 20, 21 a study of the ability to repeat nonsense words, a study of fine coordinated movement sequences of the mouth, tongue, lips and eyes in order to investigate the hypothesis of CAS/DVD. As an example, the results from the evaluation of patient 1 are available in Supplementary Data (Supplementary Table 1). Oro-facial praxis was assessed using the Henin–Dulac scale.22 Neuropsychological evaluations were conducted using the standardised Weschler’s scales. A diagnosis of CAS/DVD was made when a patient met the following criteria: (1) presence of significantly abnormal oro-facial praxis and (2) presence of five or more key features of CAS published by Forrest.23
Results
Patients 1 and 2, family 1
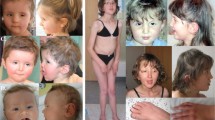
The proband (patient 1) was a 3-year-old boy, the second child born to healthy parents. His older brother had normal development and schooling. During the third trimester of pregnancy, asymmetrical cerebral ventricular dilatation was noticed (left ventricle: 11 mm). Fetal encephalic MRI, maternal CMV serology and foetal karyotype were performed and showed no additional abnormalities. At birth at 39 weeks of gestation, weight of the patient was 3270 g, length 51 cm and OFC 35 cm. Post-natal transfontanellar ultrasonography was interpreted as normal. He was described as a calm baby, with no interest in toys. Acquisition of developmental milestones was slow: walking was acquired at 21 months and speech was delayed until around the age of 3 years. He communicated mostly by shouting. He had disturbed sleep. At the first examination at 3 years of age, height and weight were on the mean curve, but he had macrocephaly at +2.5 SD. Mildly dysmorphic features were noted, including a square, coarse face, mild frontal bossing, enophtalmia, low-set ears, with anteverted and thick ear lobes, a marked philtrum, large nares, a thin upper lip and irregular and narrowly spaced teeth (Figure 1). He had hypertrichosis located on the lower region of his back. There were no other malformations at the clinical examination. Cerebral MRI, renal and cardiac ultrasounds, skeletal X-rays and fragile-X testing were unremarkable. Urinary oligosaccharides and mucopolysaccharides, assessed because of the hypertrichosis and coarse face, were normal. Audition tests were normal. At the age of 3 years, the neuropsychological evaluation identified a dysharmonic neuropsychological profile (WPPSI-III, comprehension SN 4/19, reading ability SN 8/19, cubes SN 1/19, object assembly and block design SN 11/19). The IQ could not be calculated because of the dissociation between performances and verbal abilities. Reading abilities were different from word comprehension, which was very low, and aggravated by a visual perception deficiency evidenced by difficulties in picture naming. The performances were comparable to those of children of his age. Attention deficit was noticed, but the patient was too young to confirm a diagnosis of ADHD. A specific rehabilitation programme focusing on abstraction and perception abilities was prescribed to improve adaptation. At 46 months, a speech evaluation diagnosed speech impairment with no communication troubles. Comprehension skills were low. Spontaneous speech associated with unarticulated words was understood and translated by the family. A neuropediatric examination ruled out a neurological defect. Array CGH diagnosed a 3.2-Mb telomeric deletion of the short arm of chromosome 12 (arr 12p13.33p13.32 (179 123–3 264 542) x1). qPCR analysis in the parents revealed that the deletion was inherited from the mother (patient 2).

Photos of patients 1, 3 and 9. Note the mild non-specific dysmorphic features. Patient 1: macrocephaly, square and coarse face with large forehead, enophtalmia, down-slanting palpebral fissures, low-set ears, anteverted and thick ear lobes, a marked philtrum, large nares, a thin upper lip and irregular and narrowly spaced teeth; patient 3: no dysmorphic features; and patient 9: hypotelorism, horizontal eyebrows, up-slanting palpebral fissures, large nares and downturned corner of the mouth. Photos published with parental consent.
The mother (patient 2) had a past history of severe speech delay with speech production troubles. She said her first words at 4 years of age and had intensive speech therapy. She had difficulties during her schooling, but did not attend a school for special needs. She was not able to obtain a high school diploma, but followed a vocational course. She is now working as a home-care provider and has good social integration. Her IQ is 89 (WAIS-III. IVQ: 86 with VCI 92, WMI: 94, IPQ: 96 with OPI: 95 and PSI: 102). The size of the deletion was determined by array CGH and found to be identical in patient 1 and his mother. Further investigations of the family showed that the deletion in the mother was de novo.
Specialised speech evaluation revealed several characteristic signs of CAS/DVD, including better receptive than expressive abilities, inconsistent errors on consonants and vowels in repeated production of syllables or words with an increase in errors with longer words, especially over two syllables, impaired ability to repeat nonsense words and oral apraxia in fine coordinated movement sequences of the mouth, tongue, lips and eyes. As an example, we phonetically translated an evaluation in the Supplementary Data (Supplementary File 1). A similar pattern was found in her child when testing was possible (Table 2).
Patients 3, 4 and 5, family 2
Patient 3 was the only child of young healthy non-consanguineous parents. The pregnancy was uneventful. He was born at 40 weeks of gestation. His birth weight was 2800 g, length 47 cm and OFC 34 cm. He was able to walk at 13 months. Speech was delayed with the first sentences pronounced at 3½ years. Hearing tests were normal. Behavioural abnormalities were noted at 3 years of age with hyperactivity, anxiety, solitariness and low social interaction. Several stereotypies were noticed in response to stress. There was no sleep disturbance. Standard schooling was interrupted before the child started primary school, and it was recommended that he attend a school for special-needs children. When he was referred for a neuropediatric consultation at 5 years of age, measurements were above normal with a height of 118 cm (+2.1 DS), a weight of 21 kg (+2.2 DS) and OFC 53 cm (+1.2 DS). No dysmorphic features were noted (Figure 1). Expressive speech remained insufficient with speech production trouble. The Henin–Dulac test revealed praxis troubles (Z-scores between −4 and −7 SD) associated with dysphasic symptoms (Table 2). Standard karyotyping was normal, fragile X was ruled out. Array-CGH 150K identified a 1.3-Mb deletion on the short arm of chromosome 12 (arr 12p13.33 (192 403–1 346 471) x1). This result was checked by FISH analysis using BAC probes (BAC RP11-359B12). Familial studies revealed that the deletion was inherited from the father and the paternal grandfather. Incomplete penetrance was first suggested, but targeted medical interviews suggested variable expressivity. Indeed, the father (patient 4) had a past history of speech delay with severe speech production troubles, stammering and unarticulated words. He had difficulty learning how to read and write. Hyperactivity was noticed in infancy and he had to stop school before the 7th grade at 15 years of age. He is now working as a technical operator. The paternal grandfather (patient 5) had a similar past history. The age at first words could not be defined, but he still displays speech production troubles with many unarticulated words. He was also hyperactive and stopped school before high school at 17 years of age. He has borderline intelligence and used to work as a factory worker. Clinical examinations of the three patients were normal. No dysmorphism was noticed in patients 4 and 5.
Patient 6, family 3
The proband was one of non-identical twins born to a healthy Caucasian couple following assisted conception by intracytoplasmic sperm injection. He was delivered by Caesarean section at 28 weeks of gestation due to premature rupture of the membranes and cord prolapse. His birth weight was 1320 g. He was ventilated for 2 days and then on continuous positive airway pressure for several weeks. He remained in a neonatal intensive care unit for 8 weeks where he had problems with jaundice and anaemia but no major collapses. He passed his neonatal hearing test and started feeding well. He smiled at 18 weeks, sat between 6 and 9 months and walked at 14 months but was uncoordinated. Developmental delay with late speech was noticed and there was concern that he had some autistic features. His general health was good. When he was seen at 3 years, he was able to say a dozen words with first associated words. There was no neurological defect identified during clinical examination. Most of his problems were in the area of speech and language. Speech evaluation was limited by ID and concentration troubles. Although delayed, comprehensive skills were satisfactory and expressive skills were lower, the therapist noticed very slow progress in therapy and inability in repeating words before the age of 10 years. Oro-facial praxis was not acquired and was associated with fine psychomotor difficulties (Table 2). His height was 98.7 cm (75th centile), weight 16.14 kg (75th centile) and OFC 52 cm (50th centile). He had rather myopathic facies with a tented upper lip and a tendency to drool. He snored excessively at night. His palate was highly arched. He had mild hypotonia with generalised joint laxity and an umbilical hernia. His ear lobes were prominent. The routine karyotype, 22q11 FISH, fragile X and 11p methylation were normal, as was a brain MRI scan. By 5 years of age, his speech was improving. He was declared to have special educational needs and receives extra help at school. He was in good health, and the main concern was his behaviour. He did not mix well with other children, had poor communication skills, was not motivated to learn and was late with toilet training. A diagnosis of autism was suggested but not formally confirmed. Unfortunately, he could not be evaluated by a specialised speech therapist. An SNP array study using the Affymetrix SNP6.0 array showed a 3.1-Mb deletion of 12p13.33, which was confirmed on FISH studies using the BAC probe RP11-359B12. The deletion was not present in either parent. His twin brother was developing normally.
Patient 7, family 4
The proband was the first child of young healthy parents. A younger sister aged 11 years had normal psychomotor development and schooling. The pregnancy was unremarkable and the patient was born at 40 weeks of gestation with a birth weight of 2820 g, birth length 47 cm and OFC 34 cm. There was no problem during the neonatal period. Crawling was acquired at 18 months. The first symptom was a delay with walking, still not acquired at 22 months. Later, speech delay was also noticed. The first words were pronounced at 3½ years. At 5 years of age , height was 113 cm (+1 DS), weight 7.3 kg (−1 DS) and OFC 47.5 cm (−3.5 DS). Clinical examination revealed a long face with large ears and prominent lobes, epicanthus and large incisors with dental malocclusion. Anxiety and attention-deficit hyperactivity disorder was diagnosed. Unfortunately, he could not be evaluated by a specialised speech therapist. Complementary investigations included a normal brain MRI, normal karyotype, fragile X, plasma amino-acid and urinary organic-acid chromatography and transferrin isoelectrical focusing. The array CGH identified a de novo 2.76-Mb deletion in the 12p13.33 band (arr 12p13.3 (1 080 000–3 850 000) x1). This deletion was confirmed by FISH analysis and not present on the parental chromosomes using BAC probes (RP5-927J10 and RP11-476M19).
Patient 8, family 5
The proband was a 10-year-old child of young non-consanguineous parents of Indian origin. His younger brother was healthy. A maternal uncle had low academic skills. The pregnancy was normal but complicated with a premature rupture of membranes at 31 weeks of gestation. The patient was born at 32 weeks with a birth weight of 1.9 kg. The neonatal period was complicated by hospitalisation in an intensive care unit for hypothermia and feeding difficulties. Psychomotor development was delayed with independent walking acquired at around 30 months and poor fine motor skills. The first words were pronounced at 36 months. Abnormal speech production was noticed and speech therapy was quickly started. At 9 years of age, measurements were 125 cm for height (25e percentile) and 20 kg for weight (3e percentile). There was relatively mild microcephaly with an OFC of 49.5 cm (3e percentile). Mild non-specific dysmorphism was noticed with micrognathia and prominent ears. He had surgery for inguinal hernia at 9 years of age. The patient went to a normal school with occupational therapy until the age of 10 years and was then after-orientated to a school for special-needs children. He was described as a fearful child with poor concentration skills. A diagnosis of ADHD was raised after a neuropsychological evaluation. Ritalin was tried then stopped because of a lack of effectiveness. This evaluation also noted a marked delay in arithmetic problem solving, sub-normal skills in reading and normal skills in spelling. Unfortunately, he could not be evaluated by a specialised speech therapist. Additional investigations included normal haemoglobin electrophoresis, 22q11 FISH, fragile X, plasma and urine amino acids, urine organic acids, brain MRI and EEG. Blood TSH analysis revealed moderate hypothyroidism, treated with L-thyroxine. Array CGH identified a 2.5-Mb deletion on the short arm of chromosome 12 (arr 12p13.33 (33 879–2 537 524) x1). The deletion was not found in either parent.
Patient 9, family 6
The patient was a 4-year-old child presenting initially with speech delay, contrasting with normal motor skills. Pregnancy was marked by intra-uterine growth retardation. The mother had a personal history of speech delay with no consequences on her education or professional career. For the patient, sitting was acquired at 7 months and walking at 16 months. Speech was delayed and the first words were uttered between 36 and 40 months. Clinical examination was normal. The morphological examination revealed hypotelorism, microcephaly with a prominent metopic suture, moderate joint laxity and brittle first toe nails. Chronic otitis was diagnosed and grommets were implanted. The patient was orientated to a school for special-needs children at the age of 6 years because of developmental delay associated with behavioural abnormalities. Speech evaluation identified speech production troubles contrasting with better lexical acquisition. The standardised speech evaluation identified oro-facial apraxia, associated with poor intelligibility of speech. He presented with frequent sound omissions and vowel errors (Table 2). Speech was only understood by the parents. The first sentences were noticed at the age of 8 years. Poor fine motor skills were also noticed. At the age of 11 years, growth was normal (height +1 SD and weight +1 SD). Microcephaly was persistent (OFC −2.2 SD). Cerebral MRI was normal. Standard chromosomal analysis was normal. Subtelomeric FISH analysis diagnosed a terminal deletion of the short arm of chromosome 12. Parental analysis revealed that the patient had a de novo deletion. Mapping was refined by array CGH 4 × 180k (Agilent Technologies) and identified a de novo 4.76-Mb deletion (arr 12p13.32p13.33 (163 393–4 790 279) x1).
Discussion
Subtelomeric deletions associated with developmental disabilities account for 2.5% of the etiologies of learning disability. Approximately half of the clinically significant abnormalities identified are isolated terminal deletions, the majority of which are de novo. In the largest study, which investigated the telomeres of 11 688 individuals with developmental disabilities, the 12p subtelomeric deletion was the least frequently encountered variation.24
12p13.33 microdeletion is a rare condition that has only been described in four case reports (three subtelomeric and one interstitial).25, 26, 27, 28 It is associated with ID and various psychiatric manifestations (Table 1). The age at which the first words are uttered was delayed in 2/4 patients, but no specific data regarding speech production troubles have been reported. Larger 12p terminal deletions identified by conventional cytogenetic techniques have been described in 12 patients since 1975.29, 30, 31, 32 A recognisable ‘12p deletion syndrome’ was once suggested but ruled out because of a lack of specificity.31 From our patients and the review of the literature, we have further delineated the clinical phenotype of 12p13.3 microdeletions (Table 1). Pregnancies were generally unremarkable, except for one case with unilateral ventricular dilatation and one with intra-uterine growth retardation. No malformations were found except for one case associated with oculo-auriculo-vertebral spectrum, which may have been coincidental.26 Absent to non-specific dysmorphic features were noted, but prominent ear lobes seem to be a frequent feature. The neonatal period was in general unremarkable, and in most of the cases, the first or unique symptom was the speech delay, with the first words uttered at around 36–40 months, associated or not with a delay in walking acquisition. Interestingly, speech production troubles were identified in 8/9 patients of our series (Table 1), and this feature could have oriented clinicians towards this diagnosis. The cause of the speech production troubles was CAS/DVD in investigated cases (5/9). Patients presented an association of oro-facial praxis defects and dysphasic symptoms (Table 2). They were mostly unable to imitate sounds and had better skills on automatic than imitated speech, had difficulty with articulatory movements for speech, gross and fine motor difficulties, with general oral-motor difficulties, effortful productions, groping and increased errors with increased utterance length, frequent sound omissions, prosodic irregularities and slow progress in correcting deficits in therapy (Table 2). The diagnosis of CAS/DVD was made when patients presented five or more characteristic symptoms reported in the ASHA technical report or by Forrest.4, 23 A neuropediatric examination could also rule out a neurological deficit. The association with behavioural abnormalities and developmental delay could lead to the classification of ‘apraxia of speech with complex neurobehavioral disorder’.4
Although intellectual deficiency was first suspected in most of the reported patients (Table 1), it was ruled out in some of them following a neuropsychological evaluation. As an example, the neuropsychological evaluation of patient 1 in this study revealed dissociation between low verbal and normal non-verbal performances; it was therefore not possible to calculate the total IQ. We went back to some of the authors of previously reported cases for additional information (Table 1). When our data were pooled with other reported cases, it was interesting to note that the deletion was inherited from one parent in four of the nine probands. In each case, the inheritance of the deletion was first an unexpected finding because the parent had a normal life with a job and family. Retrospective targeted interviews revealed that they all had speech delay and learning difficulties during childhood. None of them graduated from high school and they all had jobs that did not require qualifications, which favours variable expressivity rather than incomplete penetrance. When tested, they displayed a normal IQ (family 1).
These data give further examples that go against the general view that subtelomeric imbalances will lead to MCA/MR, because apparently phenotypically normal individuals can also carry subtelomeric aberrations.24 Ravnan et al24 showed that the majority of terminal deletions were found to be de novo (48/60 familial studies). The remaining cases were inherited from a single parent carrying the same deletion, the majority of whom had been reported to be phenotypically normal by the referring physician (10/12). Therefore, parental FISH studies were recommended for all patients in whom a subtelomeric rearrangement was found. Similarly, Balikova et al.33 reported subtelomeric copy-number changes in 12 families, in which the imbalance was inherited from a phenotypically normal parent (subtelomeric 2q, 3p, 4p, 4q, 6q, 10q, 17p, 17q, Xp and Yq deletions, 1q, 4q, 10q and 11q duplications). A careful clinical history and neuropsychological investigations are therefore needed in so-called asymptomatic patients. At the present time, there are a number of hypotheses to explain the variability of clinical expression encountered in these microdeletional syndromes. These include variations in genetic background, epigenetic phenomena like imprinting, expression or regulatory variation among genes in the rearrangement region and the unmasking of recessive variants residing in the single remaining allele.
From these nine new patients with a 12p subtelomeric microdeletion, we tried to define the smallest region of overlap for the speech abnormalities. The 12p subtelomeric region is not a gene-rich region with approximately 35 genes spanning the four telomeric megabases (Figure 2). Four of these genes are known to be implicated in either neuronal exchange (IQSEC3 and ELKS/ERC1),34, 35 or psychiatric disease and intellectual disabilities (SLC6A13 and CACNA1C).36, 37 The molecular data from our group of patients added to other observations in the literature allowed us to identify a small region of overlap of 260 kb containing the ELKS/ERC1 gene. This gene seems to be the best candidate for the speech sound disorder in the 12p13.33 region. The ELKS protein is not brain specific.38 In neuron cells, the ELKS protein is concentrated in the pre-synaptic active zone.35, 38 It was shown to be necessary for vesicular exocytosis in various cell types.35, 38, 39 This protein was shown to be expressed at neuro-muscular junctions,39 raising the hypothesis of altered fine cortical control of vocalisation muscles.

Alignment of the deletions identified with array CGH. The first four megabases from the telomere comprise 35 genes. Four of them are known genes expressed in the brain and implicated in neurotransmission (IQSEC3 and ELKS/ERC1) or have been assigned to psychotic phenotypes (SLC6A13 and CACNA1C). The smallest region of overlap in all the patients with speech delay (260 kb) only contains the ELKS/ERC1 gene.
Monogenic causes of CAS are rare.3 CAS have been described occasionally with genetic disorders such as galactosemia, type 1 neurofibromatosis or in chromosomal rearrangements, in particular encompassing the FOXP1 or FOXP2 genes.40, 41, 42, 43 Four genes have been reported in association with speech disorders, often associated with other neurobehavioural abnormalities. These include mutations in the CNTNAP2, FOXP1 FOXP2 and SRPX2 genes.13, 14, 15, 16, 17, 42, 43 In this series, 7/9 patients presented with behavioural troubles and 6 of them with ADHD, which could be explained by hemizygosity of ELKS/ECR1, located in the smallest region of overlap, given the similarity in the patients’ phenotype. However, other genes expressed in the brain comprised in the microdeletion, such as CACNA1C, could also in part explain the phenotype.44, 45
To conclude, 12p13.33 subtelomeric microdeletion is a rare genetic rearrangement that predisposes patients to speech sound disorders that could be defined as CAS/DVD when evaluated by a speech therapist. The ERC1/ELKS gene found in the smallest region of overlap could be a good candidate gene for CAS/DVD.
References
Tomblin JB, Records NL, Buckwalter P, Zhang X, Smith E, O’Brien M : Prevalence of specific language impairment in kindergarten children. J Speech Lang Hear Res 1997; 40: 1245–1260.
Newbury DF, Bishop DV, Monaco AP : Genetic influences on language impairment and phonological short-term memory. Trends Cogn Sci 2005; 9: 528–534.
Kang C, Drayna D : Annu Rev Genomics Hum Genet 2011; 12: 5.1–5.20.
ASHA, American Speech-Language and Hearing Association: Childhood apraxia of speech (technical report). Rockville, MD: ASHA, 2007, p 1–74.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP : A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 2001; 413: 519–523.
MacDermot KD, Bonora E, Sykes N et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet 2005; 76: 1074–1080.
Feuk L, Kalervo A, Lipsanen-Nyman M et al. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet 2006; 79: 965–972.
Shriberg LD, Ballard KJ, Tomblin JB, Duffy JR, Odell KH, Williams CA : Speech prosody, and voice characteristics of a mother and daughter with a 7;13 translocation affecting FOXP2. J Speech Lang Hear Res 2006; 49: 500–525.
Zeesman S, Nowaczyk MJ, Teshima I et al. Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. Am J Med Genet A 2006; 140: 509–514.
Fisher SE, Scharff C : FOXP2 as a molecular window into speech and language. Trends Genet 2009; 25: 166–177.
Vargha-Khadem F, Gadian DG, Copp A, Mishkin M : FOXP2 and the neuro anatomy of speech and language. Nat Rev Neurosci 2005; 6: 131–138.
Conti-Ramsden G, Simkin Z, Botting N : The prevalence of autistic spectrum disorders in adolescents with a history of specific language impairment (SLI). J Child Psychol Psychiatry 2006; 47: 621–628.
Vernes SC, Newbury DF, Abrahams BS et al2008 A functional genetic link between distinct developmental language disorders. N Engl J Med 359: 2337–2345.
O'Roak BJ, Deriziotis P, Lee C et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Pariani MJ, Spencer A, Graham JM, Rimoin DL : A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur J Med Genet 2009; 52: 123–127.
Carr CW, Moreno-De-Luca D, Parker C et al. Chiari I malformation, delayed gross motor skills, severe speech delay, and epileptiform discharges in a child with FOXP1 haploinsufficiency. Eur J Hum Genet 2010; 18: 1216–1220.
Roll P, Rudolf G, Pereira S et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet 2006; 15: 1195–1207.
Masurel-Paulet A, Andrieux J, Callier P et al. Delineation of 15q13.3 microdeletions. Clin Genet 2010; 78: 149–161.
BALE, Bilan Analytique du Langage Ecrit, Jacquier-Roux and colleagues: Laboratoire Cognisciences et Apprentissages. Grenoble, France: IUFM-Grenoble, 1999.
ELO (Examen du Langage Oral): Khomsi A. Paris: ECPA, 2001.
Alla F, Guillemin F, Colombo MC, Roy B, Maeder C : Diagnostic value of ERTL4: a screening test of language disorders in 4-year-old children. Arch Pediatr 1998; 5: 1082–1088.
Hénin N : Hénin-Dulac test. Les cahiers d’ORL 1980; 15: 809–851.
Forrest K : Diagnostic criteria of developmental apraxia of speech used by clinical speech-language pathologists. Am J Speech Lang Pathol 2003; 12: 376–380.
Ravnan JB, Tepperberg JH, Papenhausen P et al. Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet 2006; 43: 478–489.
Baker E, Hinton L, Callen DF, Haan EA, Dobbie A, Sutherland GR : A familial cryptic subtelomeric deletion 12p with variable phenotypic effect. Clin Genet 2002; 61: 198–201.
Rooryck C, Stef M, Burgelin I et al. 2.3 Mb terminal deletion in 12p13.33 associated with oculoauriculovertebral spectrum and evaluation of WNT5B as a candidate gene. Eur J Med Genet 2009; 52: 446–449.
MacDonald AH, Rodríguez L, Aceña I et al. Subtelomeric deletion of 12p: description of a third case and review. Am J Med Genet A 2010; 152: 1561–1566.
Abdelmoity AT, Hall JJ, Bittel DC et al. 1.39 Mb inherited interstitial deletion in 12p13.33 associated with developmental delay. Eur J Med Genet 2011; 54: 198–203.
Orye E, Craen M : Short arm deletion of chromosome 12: report of two new cases. Humangenetik 1975; 28: 335–342.
Kivlin JD, Fineman RM, Williams MS : Phenotypic variation in the del(12p) syndrome. Am J Med Genet 1985; 22: 769–779.
Romain DR, Goldsmith J, Columbano-Green LM, Chapman CJ, Smythe RH, Parfitt RG : Partial monosomy 12p13.1----13.3. J Med Genet 1987; 24: 434–436.
Baroncini A, Avellini C, Neri C, Forabosco A : Distal 12p deletion in a stillborn infant. Am J Med Genet 1990; 36: 358–360.
Balikova I, Menten B, de Ravel T et al. Subtelomeric imbalances in phenotypically normal individuals. Hum Mutat 2007; 28: 958–967.
Fukaya M, Kamata A, Hara Y et al. SynArfGEF is a guanine nucleotide exchange factor for Arf6 and localizes preferentially at post-synaptic specializations of inhibitory synapses. J Neurochem 2011; 116: 1122–1137.
Hida Y, Ohtsuka T : CAST and ELKS proteins: structural and functional determinants of the presynaptic active zone. J Biochem 2010; 148: 131–137.
Saus E, Brunet A, Armengol L et al. Comprehensive copy number variant (CNV) analysis of neuronal pathways genes in psychiatric disorders identifies rare variants within patients. J Psychiatr Res 2010; 44: 971–978.
Green EK, Grozeva D, Jones I et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 2010; 15: 1016–1022.
Ohara-Imaizumi M, Ohtsuka T, Matsushima S et al. ELKS, a protein structurally related to the active zone-associated protein CAST, is expressed in pancreatic beta cells and functions in insulin exocytosis: interaction of ELKS with exocytotic machinery analyzed by total internal reflection fluorescence microscopy. Mol Biol Cell 2005; 16: 3289–3300.
Grigoriev I, Yu KL, Martinez-Sanchez E et al. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol 2011; 21: 967–974.
Shriberg LD, Paul R, Black LM, Van Santen JP : The hypothesis of apraxia of speech in children with autism spectrum disorder. J Autism Dev Disord 2011; 41: 405–426.
Bishop DV : Which neurodevelopmental disorders get researched and why? PLoS One 2010; 5: e15112.
Palka C, Alfonsi M, Mohn A et al. Mosaic 7q31 deletion involving FOXP2 gene associated with language impairment. Pediatrics 2012; 129: 183–188.
Hamdan FF, Daoud H, Rochefort D et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet 2010; 87: 671–678.
Splawski I, Timothy KW, Priori SG, Napolitano C, Bloise R : Gene Reviews. Seattle: University of Washington, 1993–2006.
Thimm M, Kircher T, Kellermann T et al. Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med 2010; 16: 1–11.
Acknowledgements
We thank the Direction Générale de l'Organisation des Soins (DGOS) for their support for the development of the array-CGH platform in France, as well as the DECIPHER consortium. We also thank the Regional Council of Burgundy for their financial support, as well as families.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Thevenon, J., Callier, P., Andrieux, J. et al. 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur J Hum Genet 21, 82–88 (2013). https://doi.org/10.1038/ejhg.2012.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.116
Keywords
This article is cited by
-
Novel FOXP2 variant associated with speech and language dysfunction in a Chinese family and literature review
Journal of Applied Genetics (2024)
-
Genetic Aspects of Speech Disorders in Children
Neuroscience and Behavioral Physiology (2024)
-
Contributions of common genetic variants to specific languages and to when a language is learned
Scientific Reports (2022)
-
Non-coding structural variation differentially impacts attention-deficit hyperactivity disorder (ADHD) gene networks in African American vs Caucasian children
Scientific Reports (2020)
-
Genetic Intersections of Language and Neuropsychiatric Conditions
Current Psychiatry Reports (2020)
