
Abstract
Patients with an interstitial 13q deletion that contains the RB1 gene show retinoblastoma and variable clinical features. Relationship between phenotypic expression and loss of specific neighboring genes are unresolved, yet. We obtained clinical, cytogenetic and molecular data in 63 patients with an interstitial 13q deletion involving RB1. Whole-genome array analysis or customized high-resolution array analysis for 13q14.11q14.3 was performed in 38 patients, and cytogenetic analysis was performed in 54 patients. Deletion sizes ranged between 4.2 kb and more than 33.43 Mb; breakpoints were non-recurrent. Sequence analysis of deletion junctions in five patients revealed microhomology and insertion of 2–34 base pairs suggestive of non-homologous end joining. Milder phenotypic expression of retinoblastoma was observed in patients with deletions larger than 1 Mb, which contained the MED4 gene. Clinical features were compared between patients with small (within 13q14), medium (within 13q12.3q21.2) and large (within 13q12q31.2) deletions. Patients with a small deletion can show macrocephaly, tall stature, obesity, motor and/or speech delay. Patients with a medium deletion show characteristic facial features, mild to moderate psychomotor delay, short stature and microcephaly. Patients with a large deletion have characteristic craniofacial dysmorphism, short stature, microcephaly, mild to severe psychomotor delay, hypotonia, constipation and feeding problems. Additional features included deafness, seizures and brain and heart anomalies. We found no correlation between clinical features and parental origin of the deletion. Our data suggest that hemizygous loss of NUFIP1 and PCDH8 may contribute to psychomotor delay, deletion of MTLR1 to microcephaly and loss of EDNRB to feeding difficulties and deafness.
Similar content being viewed by others


Introduction
Retinoblastoma (Rb) is caused by mutational inactivation of the RB1 gene, a tumor suppressor located on chromosome 13q14.2. About 5–15% of the patients with Rb are heterozygous for a gross deletion that includes the entire or substantial parts of RB1.1 It has been reported that the proportion of patients with unilateral Rb in carriers of 13q deletions is higher compared with patients with intragenic loss-of-function mutations.1, 2, 3
In addition to Rb, patients with a 13q deletion involving the region 13q14.2 often present with pleiotropic features. On the basis of karyotype–phenotype associations, a classification for patients with a 13q deletion with and without Rb was proposed.4 Patients with a deletion proximal to 13q32 (group 1) show mild to moderate mental retardation, variable dysmorphic features and growth retardation. Patients with deletions extending into 13q32 (group 2) show one or more major malformations including severe microcephaly, and malformations of the brain, genitourinary and gastrointestinal tract. Group 3 comprises patients with distal deletions involving 13q33q34. The facial and neurological phenotype in patients with Rb and a 13q deletion was described first in three Japanese patients by Motegi et al.5 These patients show prominent eyebrows, a broad nasal bridge, a bulbous tip of the nose, a large mouth, a thin upper lip and a long philtrum. Baud et al6 described a series of 22 Rb patients with the most prominent features being anteverted ear lobes, a high and broad forehead, a prominent philtrum and severe mental retardation and/or motor impairment. In a study by Bojinova et al,7 frequent features included frontal bossing, a deeply grooved and long philtrum, a depressed and broad nasal bridge, a bulbous tip of the nose, a thick lower lip, a thin upper lip, broad cheeks and large ears and lobules. Additional case reports on patients with an interstitial 13q deletion involving band 13q14, describe macrocephaly, hypertelorism, proptosis, cleft palate, macroglossia, hypotonia and mild to severe developmental delay.8, 9, 10 In two patients with Rb and an interstitial deletion extending to 13q22, Hirschsprung disease was reported.11, 12 All these patients were analyzed using standard cytogenetic analysis. To date, only five patients with an interstitial 13q deletion involving the region 13q14.2 defined by array-based analyses have been reported.13, 14, 15 Caselli et al14 reported on one patient with normal clinical features and a small 1.7-Mb deletion, and two other patients with larger deletions of 19–45 Mb who showed variable clinical features including craniofacial dysmorphism, psychomotor delay, hypotonia, short stature and anomalies of feet and brain. A correlation of the extent of the deletion to the facial phenotype and other clinical features in patients with an interstitial 13q deletion proximal to the region 13q32 (group 1 of Brown's classification) is still wanting. It is also still unknown whether specific genes in the region account for specific aspects of the phenotype seen in Rb patients with an interstitial 13q deletion.
In this study, we report on 63 individuals with isolated or familial Rb who carry an interstitial 13q deletion involving RB1. To define genotype–phenotype correlations and to contribute to a functional gene map in the region, cytogenetic analysis in 54 patients was compared with array CGH analysis in 38 patients. Breakpoints were sequenced in five unrelated patients to analyze for the underlying deletion mechanism.
Materials and methods
Patients
A total of 63 Rb patients from 55 families were ascertained through the Rb outpatient clinic and Rb lab (Essen). All patients carried an interstitial 13q deletion involving RB1 with at least one breakpoint outside of RB1. Deletions had been identified during routine genetic testing using microsatellite analysis of short tandem repeat (STR) loci within RB1, quantitative multiplex PCR,1 multiplex ligation-dependent probe amplification (MLPA) (kit P047, MRC Holland, Amsterdam, Netherlands) or standard cytogenetic analysis. Informed consent for study participation was obtained. DNA from peripheral blood lymphocytes and tumor tissue was prepared by following the standard procedures.
Cytogenetic analysis
Standard cytogenetic analysis was performed in 54 patients on cultured lymphocytes with G-banding techniques and a resolution of 500–550 bands per haploid genome.
Array CGH analysis
Custom-made high-resolution oligonucleotide CGH Microarray Kit 4x44K (Agilent Technologies, Santa Clara, CA, USA) was used to map deletion breakpoints in the region 13q14.11q14.3. Whole-genome array CGH analysis was performed in 15 patients with larger deletions, using the Affymetrix 250K Nsp Array (Affymetrix, Santa Clara, CA, USA) or the Agilent Human genome CGH Microarray Kit 244K (Agilent Technologies) according to the manufacturer's instructions.
Molecular characterization of breakpoint sequences
To sequence deletion breakpoints in five patients, Long-Range PCR was performed using expanded long-template PCR system (Roche, Mannheim, Germany). Primers (Biomers, Ulm, Germany) were designed for each patient to bind upstream and downstream of the deleted segments as mapped by high-resolution array CGH. PCR was performed according to the manufacturer's instructions. In patients 21 and 26, fragments were cloned into a PCR II vector by using the TOPO TA kit (Invitrogen, Montreal, QC, Canada). In patients 26, 34 and 47, re-PCR with nested primers was performed. Sequence analysis was performed on a 3100 Genetic Analyzer (Applied Biosystems, Darmstadt, Germany).
Detection of parental origin of interstitial 13q deletions
If parental DNA was available, parental origin of the deletion was determined by genotyping of DNA polymorphisms within RB1. Three STR loci were analyzed: RBi2 (D13S153), located in intron 2 of RB1,16 RB1.20, located in intron 20,17 and a CA-repeat located at −890 bp upstream of L11910.
In patients uninformative for the above markers and in families in which no parental DNA was available (patients 4, 19, 22, 26, 43 51, 54, 55, 56, 66, 72, 73), parental origin of the deletion was determined by analysis of the methylation status of a differentially methylated CpG-island in intron 2 of RB1 using methylation-specific PCR.18
Statistical analysis
Statistical analysis including contingency analysis of association between genotype and phenotype and one-way analysis of variance was performed using JMP software (http://www.jmp.com, SAS Institute, Cary, NC, USA) and Stata 11.1 (StataCorp LP, College Station, TX, USA).
Results
Cytogenetic analysis
Conventional cytogenetic analysis showed a normal karyotype in 17 patients, a small deletion in 13q14.1q14.3 in 11 patients and an interstitial 13q deletion in 13q12.3q31.2 involving 13q14.2 in 26 patients (Table 1).
CGH array analysis
Results of array CGH analysis in 38 patients including nine Rb families are summarized in Table 1. Size and location of the deletions are shown in Figure 1. Nine patients carried a deletion with one breakpoint within RB1 (patients 17, 21, 26, 34, 47, 49, 50, 56, 58). In only two patients, 14 and 18, analysis revealed recurrence of the breakpoint region.

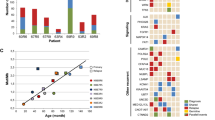
Gene map of deletions in 38 patients and cases reported in the literature. Results of array CGH analysis were uploaded into UCSC Genome browser (on the basis of NCBI Build 36.1 March 2006, hg 18).
For analysis of genotype–phenotype associations, we categorized deletions according to size to better compare clinical features. Deletions within 13q14 and smaller than 6 Mb or normal karyotype were considered to be small deletions (27 patients, including 14 patients from five families). Deletions within 13q12.3q21.2 and 6–20 Mb were considered as medium deletions (16 patients, including two patients from one family). All deletions larger than 20 Mb, including large cytogenetic deletions within 13q12q31.2, were classified as large deletions (20 patients, including two patients from one family).
Breakpoint sequence analysis
Results of breakpoint sequencing analysis in five patients are presented in Figure 2. Sequences were compared with the reference genomic sequence using a BLAT Search Genome (UCSC Genome Browser). In patient 47, we found a 2-bp microhomology at the breakpoint junction. The proximal breakpoint was located in intron 2 of the ITM2B gene and the distal breakpoint in intron 13 of RB1, and involved a Tigger 3b repeat (human transposable element). In patient 34, a 13-bp microhomology at the breakpoint junction was found, with the proximal breakpoint located in a non-repetitive sequence in intron 17 of RB1 and the distal breakpoint in a MER34C repeat. In patient 26, a 4-bp microhomology at the breakpoint junction was found, with the proximal breakpoint located in a L1MC4a repeat and the distal breakpoint in intron 2 of RB1 in a L1P1 repeat. In patient 21, a 4-bp microhomology was found, with the proximal breakpoint located in a L1PA6 repeat and the distal breakpoint in a non-repetitive sequence within intron 17 of RB1. In patient 78, we found a 34-bp insertion of unknown origin at the breakpoint junction. The proximal breakpoint mapped in a non-repetitive sequence in intron 12 of RB1, the distal breakpoint in an AluSq repeat.

Results of breakpoint sequencing analysis in five patients. Sequence data show proximal and distal breakpoints. Sequence similarities to reference genomic sequence are indicated by vertical bars.
Parental origin of the mutation
The human RB1 gene is imprinted.18 As this might be relevant for genotype–phenotype correlations, we determined the parental origin of the interstitial 13q deletions (Table 1). In 17/63 patients (27.0%), the deletion was present on the maternal chromosome, and in 44/63 patients (69.8%) on the paternal chromosome. In two patients, analysis was uninformative. In 43 patients with a de novo deletion, the deletion was present on the paternal (33 patients, 76.7%) or maternal (10 patients, 23.3%) chromosome.
Genotype–phenotype associations
Phenotypes of the study patients are listed in Tables 1 and 2. All patients with one breakpoint inside of RB1 had bilateral Rb. Of 54 patients with both breakpoints outside of RB1, 61.1% had bilateral Rb and 38.9% had unilateral or no Rb (Figure 3a, likelihood ratio test, P=0.018). Comparing deletion sizes and Rb phenotype, it appears that milder phenotypic expression, that is, unilateral Rb or incomplete penetrance, seems to be restricted to patients with deletions larger than ∼1 Mb (Figure 3b). Neither the two-sample Wilcoxon rank sum test nor the median test showed significance, but this may be because of poor statistical efficiency of these non-parametric tests.

Correlation of retinoblastoma phenotype with deletion size. (a) Box plot: proportions of patients with a deletion involving the whole RB1 gene (both breakpoints out of RB1) or part of RB1 (one breakpoint in RB1). (b) One-factor analysis.
Age at diagnosis of Rb was obtained in 50 patients. With the exception of patient 72, who was diagnosed with unilateral Rb at the age of 7 years, Rb was detected between 9 months and 3 years. In 43 patients with a de novo deletion, or who were the first affected family members (Supplementary Table 3), median age at diagnosis of unilateral Rb was 16 (5;18) months, and that of bilateral Rb was 9 (5;14) months. The difference between the two distributions was not significant (Wilcoxon rank sum test: z=−1.510, Prob>∣z∣=0.1311; median test: χ2=2.2750, P=0.131). Distribution of age at diagnosis in patients with bilateral and unilateral Rb who carry a maternal or paternal deletion was distinct, yet not statistically different (Kruskal–Wallis equality of population rank test: χ2=7.693 with three degrees of freedom, P=0.0527). The difference in age at diagnosis between patients with a maternal or paternal deletion was also not significant (Wilcoxon rank sum test: z=−0.167, Prob>∣z∣=0.8671; median test: χ2=0.3667, P=0.545). Age at diagnosis of Rb was similar between patients with a small, medium and large deletion (data not shown).
Patient 62 was born after 27 weeks of gestation; all other patients were born after 32 weeks of gestation. Mean gestational age was not distinct between patients with a small, medium and large deletion. In view of this homogeneity, comparison of birth measurements among all patients in this cohort is meaningful and showed a trend toward lower birth weight, length and head circumference in patients with a medium or large deletion compared with patients with a small deletion (Supplementary Figure 6a). Patients with a medium and a small deletion showed similar birth measurements. Measurements at time of examination showed a similar trend with a tendency to lower weight, short stature and microcephaly with increasing size of the deletion (likelihood ratio test, P=0.0365, P<0.001 and P<0.001, respectively). Of interest, some patients with a small or medium deletion showed obesity, tall stature and macrocephaly (Supplementary Figure 6b).
Hypotonia, motor and speech delay were present in almost all patients with a medium or large deletion (likelihood ratio test, P<.001, P<.001 and P=0.0022, respectively). Among patients with a small deletion, 6/15 patients showed mild to moderate motor and/or speech delay. Additional clinical features included recurrent infections, feeding difficulties and constipation, predominantly among patients with a medium and large deletion. In 17 patients, minor anomalies of the limbs were noted, including low-set thumbs, crowded toes, sandal gaps and flat arched feet (Supplementary Figure 5). Less frequent clinical features were seizures, deafness and brain and heart anomalies. Second tumors included acute myeloid leukemia at the age of 3 years (patient 13) and pineoblastoma WHO V at the age of 3 years (patient 40).
We found no correlation between the parental origin of the deletion and deletion size, Rb phenotype (unilateral or bilateral), body measurements and psychomotor development. Specifically, proportions of small, medium and large deletions and proportions of unilateral and bilateral Rb were similar among maternal and paternal de novo deletions (data not shown).
Discussion
We have analyzed genotype–phenotype correlations in a cohort of 63 patients with an interstitial 13q deletion involving RB1. Deletions were variable with respect to size and location of breakpoints; no recurrent breakpoints nor any cluster of breakpoints was observed. Sequence analysis of breakpoints in a subset of five patients revealed a mutational signature typical of non-homologous recombination mechanisms, such as non-homologous end joining.19, 20 Analysis of parental origin of de novo deletions showed a slight excess of deletions arising on the paternal chromosome 13; however, these results were less biased in favor of paternal chromosomes compared with new germline RB1 gene mutations on the whole.21 We found no significant effect of parental origin on phenotypic features.
Small RB1 mutations that lead to premature termination mutations almost invariably cause bilateral Rb.22 Among patients with cytogenetic deletions, bilateral Rb is considerably less frequent with reported proportions ranging from 18/27 (66%)3 to 9/22 (41%).6 These figures are in line with the proportion of bilateral affected patients in our cohort (42/63, 66%).
Recently, varying cancer predisposition depending on the size of the deletion was also recognized in patients with 17p13.1 microdeletions involving the TP53 gene.23 It was recognized that partial deletions of this gene lead to stronger cancer predisposition than full-length loss that include the first exon and intron, possibly because an aberrant function of this part of the TP53 accelerates tumorigenesis.23 In patients with 13q13 microdeletions, however, the pattern of genotype–phenotype correlation is distinct in that full-length loss of only the RB1 gene is associated with bilateral retinoblastoma as are intragenic mutations that result in loss of function due to premature termination.
It has been suggested that the increased frequency of unilateral Rb and non-penetrance in carriers of large contiguous deletions compared with patients with intragenic loss-of-function mutations is a consequence of a reduced spectrum of effective second mutations.2 In a patient with a deletion, second mutations that lead to homo- or hemizygosity, such as mitotic recombination or non-disjunction, will result in homozygous loss of all genes within the deleted region. If a patient's deletion contains a gene essential for basic cellular functions, fewer tumor foci will develop because only those second mutations will trigger tumor formation that leave the single copy of this gene intact. One would expect that deletions associated with milder phenotypic expression must exceed a minimum size to reach into a neighboring essential gene. This is in fact what we found. All deletions in patients with unilateral Rb or non-penetrant carriers are larger than 1 Mb. Further, it seems that once a deletion has exceeded this threshold of size, there will be no further reduction of tumor foci as we observe no increase of the proportion of unilateral disease in patients with very large deletions (Figure 3b).
Our data also provide clues as to the identity of the neighboring cell essential genes. All four patients (patients 15, 21, 26 and 47) with haploinsufficiency for ITM2B, but no involvement of MED4, have bilateral Rb. This finding suggests that loss of the ITM2B gene does not inhibit development of tumor foci. One familial deletion (patients 38, 39) involves ITM2B and MED4 but not SUCLA2 and is associated with unilateral Rb phenotype. Of a total of 27 deletions resulting in haploinsufficiency for ITM2B, MED4 and SUCLA2, nine deletions (33%) are associated with unilateral disease (patients 10, 14, 16, 18, 36, 41, 57, 63, family 70, 73) or no Rb (patient 60). Thus, deletions including MED4 are associated with milder phenotypic expression. MED4 (mediator of RNA polymerase II transcription, subunit 4) is ubiquitously expressed and encodes for vitamin D receptor-interacting protein (DRIP) complex that binds nuclear receptors.24 This suggests that alteration of vitamin D signaling through homozygous loss of MED4 is not tolerated by Rb precursor cells. It will be interesting to examine whether hemizygous loss of MED4 has an effect on the growth of Rb, pointing out to vitamin D signaling as a potential target for Rb therapy.
Most patients in our cohort are still young and this might be sufficient to explain why only a few second tumors were observed. Acute myelogenous leukemia, seen in patient 13, has been reported as a rare secondary malignancy among patients with Rb. Gombos et al25 identified several patients with secondary acute myelogenous leukemia in childhood, many of whom were treated with chemotherapy, as was patient 13. This supports the link between chemotherapy and acute myelogenous leukemia in children with Rb. In patient 16 the BRCA2 gene, located in 13q13.1, was also deleted. Heterozygous point mutations in BRCA2 predispose to breast and/or ovarian cancer26 but, to our knowledge, no patient with a deletion in this region and with breast cancer has been reported to date. Nevertheless, surveillance in Rb patients with deletions extending to 13q13.1 should include tumors associated with BRCA2 gene mutations.
The patients in our cohort showed variable craniofacial dysmorphism. In patients with small deletions, facial features were highly variable and nonspecific (Figure 4a). This finding contrasts previous reports by Motegi et al,5 Baud et al6 and Bojinova et al,7 who suggested that a distinctive facial phenotype is associated with a deletion of band 13q14. In patients with a medium deletion, craniofacial features included a high forehead, a short nose, a small upper lip and curly hair (Figure 4b). Patients with a large interstitial 13q deletion showed a round face, a high forehead, a short nose, a small upper lip and down-turned corners of the mouth (Figure 4c). Patients with a deletion extending to the region 13q22q31.2 showed mild hypertelorism, low-set ears and micrognathia, similar to patients reported with an interstitial deletion involving 13q22 and Hirschsprung disease or Waardenburg–Shah syndrome.27, 28, 29 Micrognathia was reported as a common dysmorphic feature in patients with 13q deletions and was associated with loss of the region 13q21.33q31.1.30 Additional mild anomalies of the feet were found in 17 patients. Caselli et al 2007 also reported on toe crowding and a short V toe in 2/3 patients with an interstitial 13q deletion.

Facial phenotype. (a) Patients with a small deletion show a high forehead, a broad nose tip and a thin upper lip. (b) Patients with a medium deletion show a high forehead, deep-set eyes, a short nose in younger children, a small upper lip and often curly hair. (c) Patients with a large deletion show a round face in younger children, a long face in adult patients, a high forehead, a short nose, a long philtrum in older patients, a small upper lip and down-turned corners of the mouth. Patients 4, 8, 11, 20, 72, 76, 77 and 81 have hypertelorism, low-set ears and micrognathia.
Microcephaly was present in 57.1% of the patients with a large deletion. Our findings suggest that the region 13q21.32q21.33 is critical for microcephaly. Eight genes are located within this critical region: PCDH9, KLHL1, ATXN8OS, DACH1, C13orf34, DIS3, PIBF1 and KLF5. A good candidate gene is PCDH9 that encodes for a cadherin-related neuronal receptor that localizes to synaptic junctions and has a putative role in specific neuronal connections and signal transduction.31 Of note, all deletions found in our cohort are centromeric to 13q33.3q34, a region that was reported to be critical for microcephaly by Kirchhoff et al.30
Interestingly, a few patients in this cohort and in previous reports showed macrocephaly.8, 9 However, the pattern of deleted regions was the same as that in patients with normocephaly. Of note, the MTLR1/MLNR gene, which encodes for the growth hormone secretagogue receptor found in the pituitary gland and brain, is involved in the control of growth hormone release,32 and is deleted in 5/6 patients with macrocephaly in our cohort.
Short stature was observed in 35.6% of the patients with a medium deletion and in 75% of the patients with a large deletion. This corresponds to findings in other patients with deletions proximal to the 13q31.4 Interestingly, two patients in our study showed tall stature (patient 15 and 73). The deletions in these two patients were small and overlapped in a small region within 13q14.2 that includes four genes, ITM2B, RB1, P2RY5 and RCBTB2.
Comparison of psychomotor development shows that 6/15 patients with a small deletion showed motor and/or speech delay. This is in contrast to previous studies that suggested that patients with small deletions limited to band 13q14 show normal neurological development during infancy.6, 7 Motor delay was seen in 85.7% of the patients with a medium deletion and all patients with a large deletion. Speech delay was also common among patients with a medium (84.6%) and a large (86.7%) deletion. A plausible candidate gene for psychomotor delay in 13q14.12 is NUFIP1, which is deleted in 17/22 patients with motor and/or speech delay. NUFIP1 interacts with FMRP, an RNA-binding protein encoded by FMR1, which is responsible for the fragile X syndrome.33 Another candidate gene located in 13q21.1 is PCDH8, which encodes for an integral membrane protein and is thought to function in cell adhesion in a CNS-specific manner.31
Constipation and feeding difficulties were frequent findings in patients with a medium (23.1%) and a large (44.4%) deletion. A candidate gene for constipation is EDNRB, a G protein-coupled receptor located in 13q22.3. Dosage-sensitive mutations in EDNRB have been associated with Hirschsprung disease type 2, pigment anomalies and hearing loss.34 Hirschsprung disease has been reported in other patients with an interstitial deletion involving the region 13q22.8, 27, 28, 35, 36 The milder form of constipation without evidence of intestinal aganglionosis seen in the reported patients may be explained by regulatory effects.
MRI or CT scan showed hypoplastic or partial aplastic corpus callosum in 3/25 patients. Corpus callosum hypoplasia has also been reported in two patients with deletions 13q13.1q21.1 and 13q14.11q31.1 mapped by array CGH.14 In the study by Rodjan et al,37 MRI scan in seven patients with a 13q deletion showed corpus callosum agenesis in one patient and a Dandy–Walker variant in another patient, but locations of the deletions were not reported. Ballarati et al13 and Kirchhoff et al30 refined the region 13q32.3q33.1 as a critical region for corpus callosum agenesis but no specific candidate gene was found. From the results in this cohort and the patients reported by Caselli et al,14 a second critical region for corpus callosum anomalies can be suspected further centromeric.
Following our data, patients with a proximal interstitial 13q deletion involving the RB1 gene present with a spectrum of characteristic clinical features that contrast the wider spectrum of major dysmorphism and severe congenital malformations in patients with a 13q deletion involving the terminal chromosomal region 13q32-qter.4, 13, 30, 38, 39, 40, 41, 42, 43, 44 Thus, analysis of the precise location and size of the deletion is needed to better inform families and physicians about the clinical expectations and survival in patients with a 13q deletion.
Further studies of candidate genes in the region around RB1 are needed to correlate gene functions to specific clinical phenotypes. Analysis of the parental origin in more patients with an interstitial 13q deletion is needed to further analyze for a possible functional relevance of RB1 imprinting.
References
Albrecht P, Ansperger-Rescher B, Schüler A, Zeschnigk M, Gallie B, Lohmann DR : Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum Mutat 2005; 26: 437–445.
Bunin GR, Emanuel BS, Meadows AT, Buckley JD, Woods WG, Hammond GD : Frequency of 13q abnormalities among 203 patients with retinoblastoma. J Natl Cancer Inst 1989; 81: 370–374.
Matsunaga E : Retinoblastoma: host resistance and 13q- chromosomal deletion. Hum Genet 1980; 56: 53–58.
Brown S, Russo J, Chitayat D, Warburton D : The 13q- syndrome: the molecular definition of a critical deletion region in band 13q32. Am J Hum Genet 1995; 57: 859–866.
Motegi T, Kaga M, Yanagawa Y et al: A recognizable pattern of the midface of retinoblastoma patients with interstitial deletion of 13q. Hum Genet 1983; 64: 160–162.
Baud O, Cormier-Daire V, Lyonnet S, Desjardins L, Turleau C, Doz F : Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet 1999; 55: 478–482.
Bojinova RI, Schorderet DF, Addor MC et al: Further delineation of the facial 13q14 deletion syndrome in 13 retinoblastoma patients. Ophthalmic Genet 2001; 22: 11–18.
Shanske A, Ferreira JC, Leonard JC, Fuller P, Marion RW : Hirschsprung disease in an infant with a contiguous gene syndrome of chromosome 13. Am J Med Genet 2001; 102: 231–236.
Skrypnyk C, Bartsch O : Retinoblastoma, pinealoma, and mild overgrowth in a boy with a deletion of RB1 and neighbor genes on chromosome 13q14. Am J Med Genet A 2004; 124: 397–401.
Slavotinek AM, Lacbawan F : Large interstitial deletion of chromosome 13q and severe short stature: clinical report and review of the literature. Clin Dysmorphol 2003; 12: 195–196.
Weigel BJ, Pierpont ME, Young TL, Mutchler SB, Neglia JP : Retinoblastoma and Hirschsprung disease in a patient with interstitial deletion of chromosome 13. Am J Med Genet 1998; 77: 285–288.
Zaborowski AG, Kruse CH, Kavonic S, Pergorano RJ : Retinoblastoma and Hirschsprung disease with a 13q14 to 22 deletion. J Pediatr Ophthalmol Strabismus 2008; 45: 366–367.
Ballarati L, Rossi E, Bonati MT et al: 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet 2007; 44: e60.
Caselli R, Speciale C, Pescucci C et al: Retinoblastoma and mental retardation microdeletion syndrome: clinical characterization and molecular dissection using array CGH. J Hum Genet 2007; 52: 535–542.
Kogan JM, Egelhoff JC, Saal HM : Interstitial deletion of 13q associated with polymicrogyria. Am J Med Genet A 2008; 146: 910–916.
Toguchida J, McGee TL, Paterson JC et al: Complete genomic sequence of the human retinoblastoma susceptibility gene. Genomics 1993; 17: 535–543.
Yandell DW, Dryja TP : Detection of DNA sequence polymorphisms by enzymatic amplification and direct genomic sequencing. Am J Hum Genet 1989; 45: 547–555.
Kanber D, Berulava T, Ammerpohl O et al: The human retinoblastoma gene is imprinted. PLoS Genet 2009; 5: e1000790.
Roth DB, Porter TN, Wilson JH : Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol 1985; 5: 2599–2607.
Shaw CJ, Lupski JR : Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 2004; 13 (Spec No 1): R57–R64.
Dryja TP, Mukai S, Petersen R, Rapaport JM, Walton D, Yandell DW : Parental origin of mutations of the retinoblastoma gene. Nature 1989; 339: 556–558.
Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B : The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am J Hum Genet 1996; 58: 940–949.
Shlien A, Baskin B, Achatz MI et al: A common molecular mechanism underlies two phenotypically distinct 17p13.1 microdeletion syndromes. Am J Hum Genet 2010; 87: 631–642.
Rachez C, Lemon BD, Suldan Z et al: Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 1999; 398: 824–828.
Gombos DS, Hungerford J, Abramson DH et al: Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology 2007; 114: 1378–1383.
Wooster R, Bignell G, Lancaster J et al: Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789–792.
Kiss P, Osztovics M : Association of 13q deletion and Hirschsprung's disease. J Med Genet 1989; 26: 793–794.
Lamont MA, Fitchett M, Dennis NR : Interstitial deletion of distal 13q associated with Hirschsprung's disease. J Med Genet 1989; 26: 100–104.
Tuysuz B, Collin A, Arapoglu M, Suyugul N : Clinical variability of Waardenburg-Shah syndrome in patients with proximal 13q deletion syndrome including the endothelin-B receptor locus. Am J Med Genet A 2009; 149A: 2290–2295.
Kirchhoff M, Bisgaard AM, Stoeva R et al: Phenotype and 244k array-CGH characterization of chromosome 13q deletions: an update of the phenotypic map of 13q21.1-qter. Am J Med Genet A 2009; 149A: 894–905.
Strehl S, Glatt K, Liu QM, Glatt H, Lalande M : Characterization of two novel protocadherins (PCDH8 and PCDH9) localized on human chromosome 13 and mouse chromosome 14. Genomics 1998; 53: 81–89.
McKee KK, Tan CP, Palyha OC et al: Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics 1997; 46: 426–434.
Bardoni B, Schenck A, Mandel JL : A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum Mol Genet 1999; 8: 2557–2566.
Puffenberger EG, Hosoda K, Washington SS et al: A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 1994; 79: 1257–1266.
Khong TY, Ford WD, Haan EA : Umbilical cord ulceration in association with intestinal atresia in a child with deletion 13q and Hirschsprung's disease. Arch Dis Child Fetal Neonatal Ed 1994; 71: F212–F213.
Van Camp G, Van Thienen MN, Handig I et al: Chromosome 13q deletion with Waardenburg syndrome: further evidence for a gene involved in neural crest function on 13q. J Med Genet 1995; 32: 531–536.
Rodjan F, de Graaf P, Moll AC et al: Brain Abnormalities on MR Imaging in Patients with Retinoblastoma. AJNR Am J Neuroradiol 2010; 31: 1385–1389.
Alanay Y, Aktas D, Utine E et al: Is Dandy-Walker malformation associated with ‘distal 13q deletion syndrome’? Findings in a fetus supporting previous observations. Am J Med Genet A 2005; 136: 265–268.
Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D : A chromosomal deletion map of human malformations. Am J Hum Genet 1998; 63: 1153–1159.
Christofolini DM, Yoshimoto M, Squire JA, Brunoni D, Melaragno MI, Carvalheira G : Hydrocephaly, penoscrotal transposition, and digital anomalies associated with de novo pseudodicentric rearranged chromosome 13 characterized by classical cytogenetic methods and mBAND analysis. Am J Med Genet A 2006; 140: 1321–1325.
Gutierrez J, Sepulveda W, Saez R, Carstens E, Sanchez J : Prenatal diagnosis of 13q- syndrome in a fetus with holoprosencephaly and thumb agenesis. Ultrasound Obstet Gynecol 2001; 17: 166–168.
Quelin C, Bendavid C, Dubourg C et al: Twelve new patients with 13q deletion syndrome: genotype-phenotype analyses in progress. Eur J Med Genet 2009; 52: 41–46.
Tranebjaerg L, Nielsen KB, Tommerup N, Warburg M, Mikkelsen M : Interstitial deletion 13q: further delineation of the syndrome by clinical and high-resolution chromosome analysis of five patients. Am J Med Genet 1988; 29: 739–753.
Walczak-Sztulpa J, Wisniewska M, Latos-Bielenska A et al: Chromosome deletions in 13q33-34: report of four patients and review of the literature. Am J Med Genet A 2008; 146: 337–342.
Acknowledgements
This study was supported by the Essener Elterninitiative zur Unterstützung krebskranker Kinder e.V. This study would not have been possible without the invaluable assistance of the patients and their families. We thank the cooperating physicians for referral of the patients to our department, especially the Ophthalmologic Department, University Hospital Essen. We also thank Birgit Ansperger-Rescher and Saskia Seeland for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Mitter, D., Ullmann, R., Muradyan, A. et al. Genotype–phenotype correlations in patients with retinoblastoma and interstitial 13q deletions. Eur J Hum Genet 19, 947–958 (2011). https://doi.org/10.1038/ejhg.2011.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.58
Keywords
This article is cited by
-
Nanotechnology-based strategies overcoming the challenges of retinoblastoma: a comprehensive overview and future perspectives
Future Journal of Pharmaceutical Sciences (2024)
-
Prenatal diagnosis of distal 13q deletion syndrome in a fetus with esophageal atresia: a case report and review of the literature
Journal of Medical Case Reports (2022)
-
Retinoblastoma and mosaic 13q deletion: a case report
International Journal of Retina and Vitreous (2021)
-
13q deletion syndrome resulting from balanced chromosomal rearrangement in father: the significance of parental karyotyping
Molecular Cytogenetics (2020)
-
A female patient with retinoblastoma and severe intellectual disability carrying an X;13 balanced translocation without rearrangement in the RB1 gene: a case report
BMC Medical Genomics (2019)

{kind=link}
{kind=link}
{kind=link}