
Abstract
Alport syndrome (ATS) is a hereditary nephropathy often associated with sensorineural hypoacusis and ocular abnormalities. Mutations in the COL4A5 gene cause X-linked ATS. Mutations in COL4A4 and COL4A3 genes have been reported in both autosomal recessive and autosomal dominant ATS. The conventional mutation screening, performed by DHPLC and/or Sanger sequencing, is time-consuming and has relatively high costs because of the absence of hot spots and to the high number of exons per gene: 51 (COL4A5), 48 (COL4A4) and 52 (COL4A3). Several months are usually necessary to complete the diagnosis, especially in cases with less informative pedigrees. To overcome these limitations, we designed a next-generation sequencing (NGS) protocol enabling simultaneous detection of all possible variants in the three genes. We used a method coupling selective amplification to the 454 Roche DNA sequencing platform (Genome Sequencer junior). The application of this technology allowed us to identify the second mutation in two ATS patients (p.Ser1147Phe in COL4A3 and p.Arg1682Trp in COL4A4) and to reconsider the diagnosis of ATS in a third patient. This study, therefore, illustrates the successful application of NGS to mutation screening of Mendelian disorders with locus heterogeneity.
Similar content being viewed by others

Introduction
Alport syndrome (ATS) is an inherited disorder of type IV collagen, the major collagenous constituent of the basement membrane (BM).1 Clinically, it presents as a progressive inherited nephropathy characterized by the association of progressive hematuric nephritis with ultrastructural changes of the glomerular basement membrane (irregular thinning, thickening and splitting), high-tone sensorineural hearing loss and ocular lesions (anterior lenticonus, macular flecks, corneal endothelial vesicles, recurrent corneal erosion and cataract).2, 3, 4 ATS accounts for 1–2% of all patients who start renal replacement therapy in Europe, with an estimated frequency of about 1 in 5000.5, 6 The disease is genetically heterogeneous, but the majority (∼ 85%) of ATS kindreds show X-linked dominant inheritance (OMIM no. 301050) and are caused by mutations in the COL4A5 gene located in the Xq22 region.7 In this form, males are more severely affected than females; 70% of affected males reach end-stage renal disease (ESRD) before the age of 30 years (juvenile form), whereas the remaining 30% progress toward ESRD after 30 years (rare adult form).6, 8, 9 Females with this form of ATS usually only have microhematuria. However, there are some cases of females as seriously affected as males.10, 11 Dominant and recessive autosomal forms of ATS (OMIM nos. 104200, 203780) have also been reported and are characterized by mutations in COL4A3 and COL4A4 genes located in 2q36–37.12, 13 In the recessive form, females are usually as severely affected as males, reaching ESRD in the first or second decade of life.14, 15, 16 Heterozygous parents may be completely asymptomatic or may have isolated microhematuria or may carry some risk of renal disease progression as in autosomal dominant ATS. This latter form has been described more recently.17, 18 Both female and male patients show high clinical variability with a renal phenotype ranging from isolated haematuria to late onset ESRD, associated, in few instances, with hearing loss.12, 17, 18, 19, 20
Considering the broad spectrum of phenotypes associated with ATS, the absence of mutational hot spots, and the large size of the genes implicated in ATS, mutational analyses by standard techniques, such as DHPLC and/or direct Sanger sequencing, represent a very expensive and exhausting molecular testing. Recent advances in DNA sequencing technology, namely next-generation sequencing (NGS), have provided a powerful new approach for a simultaneous analysis of large numbers of coding regions. We used a method coupling amplicon based gene capture with resequencing on a 454 Roche platform (Genome Sequencer Junior System, Roche Applied Science, Mannheim, Germany) to identify mutations in three Alport patients, analysing all the three genes in a single experiment. This method proved to be sensitive and fast allowing for the mutational analysis of 151 amplicons/patient and for test reporting in 6 working days.
Patients and methods
Patients selection
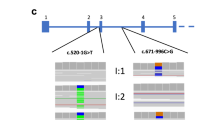
We selected three patients previously screened by DHPLC analysis.20 Patient 1 had an uncertain diagnosis of ATS and DHPLC analysis failed to identify any mutation. Patient 2 had a confident diagnosis of ATS and DHPLC analysis, followed by Sanger sequencing, detected a mutation in the COL4A3 gene (p.Gly1045Val). Patient 3 had a confident diagnosis of ATS and DHPLC analysis, followed by Sanger sequencing, identified a mutation in the COL4A4 gene (c.4749_4751delGTC). Figure 1 illustrates pedigrees of the three patients. All patients and relatives signed a written information consent. Procedures were in accordance with the Helsinki Declaration of 1975 (and as revised in 1983).

Pedigrees of families of Patients 1, 2 and 3. Gray symbols stand for microscopic hematuria and or proteinuria. Black symbols stand for ESRD.
Patient 1 (no. 3415)
This patient was 18 years old and presented an atypical clinical picture with onset of isolated persistent proteinuria at the age of 15, only one episode of gross hematuria at 18, and normal renal function. Ultrastructural analysis of a renal biopsy revealed thin glomerular basement membrane nephropathy. In his family history, only the mother presented isolated and persistent microscopic hematuria with normal renal function (Figure 1).
Patient 2 (no. 2740)
Patient 2 was 34 years old and presented a typical severe clinical picture with onset of microscopic hematuria and proteinuria at the age of 10 years (Figure 1). Since then, further clinical investigation revealed steady progression of disease until end renal stage disease at age 24. The patient underwent renal transplantation at the age of 32. Renal biopsy was not performed. Parents of the patient were unaffected and three of five siblings (two brothers and one sister) presented a similar clinical course (Figure 1). Especially in the sister, ultrastructural analysis of renal biopsy was suggestive for ATS.
Patient 3 (no. 3017)
Patient 3 was 15 years old and since 3 months of age presented microscopic hematuria and onset of proteinuria at the age of 12 years (Figure 1). He has normal renal function. Ultrastructural analysis of renal biopsy was compatible with a diagnosis of ATS. Audiometry showed bilateral high-frequency sensorineural hearing loss. His family history was positive for microscopic hematuria in paternal and maternal pedigrees; both parents presented normal renal function and microscopic hematuria (Figure 1).
Samples and DNA extraction
Genomic DNA was isolated from EDTA peripheral blood samples using a QIAamp DNA Blood Kit according to the manufacturer's protocol (Qiagen, http://www.qiagen.com).
Amplicon library preparation
To analyse sequence variations in COL4A3, COL4A4 and COL4A5 genes, we used a strategy based on the locus-specific amplification of genomic DNA, amplifying each amplicon separately, followed by Roche 454 resequencing. Fusion primers were designed to generate tiled amplicons ranging in size between 200–300 bp segments (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunior_AmpliconLibraryPrep-RevJune2010.pdf; Supplementary Table 4). At the 5′ end fusion primers contained an additional sequence, MIDs, that barcodes the sample (Supplementary Table 4). The MID sequence was selected from a list provided from Roche. Thermal cycling was performed on an Applied Biosystems 2720 Thermal Cycler using the following cycling profile: one cycle at 95 °C for 5 min followed by 35 cycles at 95 °C for 30 s, at the specific annealing temperature for 30 s, at 72 °C for 30 s, followed by a final extension step at 72 °C for 5 min (Supplementary Table 4). Small DNA fragments were removed using AMPure PCR purification system (Agencourt, Beverly, MA, USA) following the manufacturer's protocol (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunior_AmpliconLibraryPrep-RevJune2010.pdf). Amplicons were subsequently quantified using the Quant-iT PicoGreen dsDNA reagent (Invitrogen Corporation, Life Techologies, Carlsbad, CA, USA). All amplicons were then pooled at an equimolar ratio. Subsequently the sample pool was diluted to a final concentration of 1 × 107 PCR fragment molecules/μl.
GS junior sequencing
The amplicon-PCR-derived fragments were annealed to carrier beads and clonally amplified by emulsion PCR (emPCR). emPCR was performed according to the manufacturer's protocol (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunior_emPCR_Lib-A_RevApril2011.pdf). The beads were isolated and compartmentalized into droplets of an aqueous PCR reaction buffer in oil emulsion. Subsequently, the emulsions were broken by isopropanol to facilitate collection of the amplified fragments bound to their specific beads. The beads carrying single-stranded DNA templates were enriched, counted and deposited into the PicoTiterPlate for sequencing (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunior_Sequencing-MM-RevJune2010.pdf). The 454 technology is based on pyrosequencing, a sequencing approach based on chemiluminescent detection of pyrophosphate released during polymerase-mediated deoxynucleoside triphosphate incorporation.21, 22, 23 During sequencing a CCD camera-based imaging assembly was used to capture the pyrosequencing-derived light signal and to collect the readout data per flow, which was then used by a Genome Sequencer-specific base-caller to generate the sequence reads.
Data analysis
Data analysis was performed using the Roche proprietary software package for the GS Junior system. Image acquisition, image processing and signal processing were performed during the run. Post run analysis was conducted using the latest version (2.5p1) of GS Amplicon Variant Analyzer (AVA; http://454.com/downloads/my454/documentation/gs-junior/software-manual/454_Sequencing_Software_Manual_v2.5p1_PartD.pdf). The AVA application computes the alignment of reads from Amplicon libraries obtained on the GS Junior Instrument, and identifies differences between the reads and a reference sequence. In this study, amplicon nucleotide sequence reads were aligned to the Human Mar. 2006 (hg18) assembly genomic sequence of COL4A3, COL4A4 and COL4A5. The AVA software identifies all nucleotide variants, and provides read counts and frequencies (Supplementary Tables 1–3). Variations are also displayed graphically with a histogram indicating positions of variation (Figures 2 and 3). Individual flow grams were reviewed to examine and confirm all variant calls made by the software.

Flowchart illustrating the different steps to filter variations detected by 454 technology in a pilot study of three ATS patients. This approach allowed to identify pathogenic mutations and to indicate cut-off values useful for flagging false-positive results.

Patient 2 (no. 2740) mutation detection. (a) A screenshot from the GS Amplicon Variant Analyzer software showing the COL4A3 missense sequence variant c.3440C>T (p.Ser1147Phe). The upper panel corresponds to a histogram indicating the percentage of variations. In the lower panel, reads from different directions are displayed and the mutated base is shown between the two vertical blue lines. Near the variation, there is a polyT stretch that creates technical artifacts (Supplementary Table 2). (b) Sanger sequencing chromatograms showing the missense sequence variant c.3440C>T (p.Ser1147Phe) found in Patient 2 respect to a control sample. The color reproduction of this figure is available at the European Journal of Human Genetics online.
Sanger sequencing
Direct sequencing of the purified PCR products, obtained with the same primers and PCR conditions (Supplementary Table 4) described for amplicon library preparation, was performed in both directions (PE Big Dye Terminator Cycle Sequencing Kit) on an ABI Prism 310 genetic analyser (PE Applied Biosystems, Forest City, CA, USA) and analyzed with the Sequencer software.
Results
Overall analysis of variants
Sequencing output of the three probands (Figure 1) were visualized by GS Amplicon Variant Analyzer software (Roche Applied Science) in a table containing gene name, type of variant, percentage of forward and reverse sequences containing the variant with specification of the numbers of passed filter sequences (Supplementary Tables 1–3). In the table, variants could be ordered on the basis of either the percentage of sequence variants (Supplementary Tables 1–3) or the DNA position (Table 1).
After mapping, 100% of the targeted region was covered in each patient. Percentages covered at >100-fold depth were 99% in Patient 1, 94% in Patient 2 and 99% in Patient 3. The mean depth of coverage was 630 in Patient 1, 430 in Patient 2 and 595 in Patient 3. A total of 356 variants were detected, with an average of 118 variants per patient within the 35 966 bp targeted region (∼1 variant per 3 kb; Figure 2; Supplementary Tables 1–3).
In this pilot study, the gold standard Sanger method was used to distinguish real changes from false positives (Figure 2; Supplementary Tables 1–3). Sanger sequencing confirmed a mean of 22 variants per patient. To distinguish pathogenic mutations from polymorphisms, we compared results with literature and personal data and with public databases of known sequence variants (db(SNP) single nucleotide polymorphism; Figure 2; Table 1). All confirmed variants corresponded to known polymorphisms except for four variants: two already identified pathogenic mutations (p.Gly1045Val in COL4A3, patient 2 and p.Gln1583_Ser1584SdelInsHis in COL4A4, patient 3) and two new sequence variants (p.Ser1147Phe in COL4A3, patient 2 and p.Arg1682Trp in COL4A4, patient 3) not detected by previous DHPLC screening (Table 1; Figures 3 and 4).20

Patient 3 (no. 3017) mutation detection. (a) A screenshot from the GS Amplicon Variant Analyzer software showing the COL4A4 missense sequence variant c.5044C>T (p.Arg1682Trp). The upper panel corresponds to a histogram indicating the percentage of variations. In the lower panel, reads from different directions are displayed and the mutated base is shown between the two vertical blue lines. (b) Sanger sequencing chromatograms showing the missense sequence variant c.5044C>T (p.Arg1682Trp) found in Patient 3 respect to a control sample. The color reproduction of this figure is available at the European Journal of Human Genetics online.
Patient 1 (no. 3415)
A total of 25 611 080 bp represented by 104 222 reads (57.3%) with a mean read length of 245 bp passed quality filter systems provided in the GS Junior software. The remaining 42.7% did not pass the Genome Sequencer software's internal quality control parameters for various reasons: short read length (33.64%) or incomplete extension and mixed reads (9.05%). A total of 136 variants were detected by Roche 454 sequencing (Figure 2; Supplementary Table 1). Among them, 24 were confirmed by Sanger sequencing (Figure 2; Table 1). These 24 variants (15 exonic and 9 intronic) have all been reported as known polymorphisms in the literature or in personal data repositories (Figure 2; Table 1).12, 24, 25, 26, 27, 28, 29 A total of 16 variants were identified in 82–100% of the sequences (Supplementary Table 1). Sanger sequencing confirmed that these variants were present in homozygous/hemizygous state. Five variants were present in 43–66% of the sequences (Supplementary Table 1). Sanger sequencing demonstrated that these variants were present in heterozygous state. One sequence variant (IVS46-8T>C) was identified in 98% of forward sequences and 8% of reverse sequences (Supplementary Table 1). Sanger sequencing demonstrated that this was a variant in heterozygous state. Two variants identified in a high percentage of forward sequences (83 and 92%) and in a percentage of ∼50% of reverse sequences (42 and 49%) were demonstrated to be in homozygous state by Sanger sequencing (Supplementary Table 1). Four variants, identified in a significant percentage of forward (99, 98, 89 and 89%) and reverse (99, 99, 54 and 46%) sequences, were not confirmed by Sanger sequencing (Supplementary Table 1). All of these sequence changes were found in polyT stretches (Figure 2; Supplementary Table 1). Two other variants with highly unbalanced sequence results (93 and 84% of reverse sequences and none in the forward sequences) also were not confirmed by Sanger sequencing (Supplementary Table 1). These, too, were located within polyT stretches (Supplementary Table 1). This observation, in addition to Sanger sequence results, led us to interpret these variants as technical artifacts. Unbalanced results with one sequence strand detected in a significant percentage (26–71%) but the other strand in a percentage near zero (0–4%) were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 1). Variants found in a low percentage of both strands (≤20%) were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 1).
Patient 2 (no. 2740)
A total of 22 821 584 bp represented by 94 405 reads (49.18%) with a mean read length of 240 bp passed the Genome Sequencer software's internal quality control system. The remaining 51% did not pass quality filters for short read length (41.81%) or incomplete extension and mixed reads (9.01%). A total of 103 variants were detected by the GS Junior System (Figure 2; Supplementary Table 2). Among them, 23 variants were confirmed by Sanger sequencing (Figure 2; Table 1). All of these variants (16 exonic and 7 intronic) were known polymorphisms except for two in the COL4A3 gene (NM_031362): p.Gly1045Val (c.3134G>T) and p.Ser1147Phe (c.3440C>T; Figures 2 and 3; Table 1).12, 24, 25, 26, 27, 28, 29 Even if we did not obtain any sequence by NGS for the reverse strand, the first of these novel sequence variants was previously identified in heterozygous state by DHPLC analysis and Sanger sequencing.20 This variant was inherited from the healthy father (Figure 1). The second one, confirmed by Sanger sequencing, was not previously detected by DHPLC analysis and has been never reported as polymorphism (Figure 3). We determined that this variation was inherited from the healthy mother and was absent in the two healthy brothers (Figure 1). The variation was absent in a control population of 100 Italian individuals (A Renieri, unpublished data). The variation causes the substitution of the small uncharged polar serine with the aromatic nonpolar phenylalanine in the collagenous domain of the protein. Furthermore, serine at position 1147 resulted moderately conserved in different species (Supplementary Figure 1; http://genome.ucsc.edu/). However, PolyPhen (http://genetics.bwh.harvard.edu/pph) analysis, used to evaluate the likelihood that the observed variation alters the function of the protein, predicted the missense change to be likely benign.
Eight out of 22 variants were present in 85 to 100% of forward and reverse sequences and Sanger sequencing confirmed that they were present in homozygous/hemizygous state (Supplementary Table 2). A total of 14 variants were present in 27 to 59% of forward and reverse sequences and Sanger sequencing demonstrated that these variants were present in heterozygous state (Supplementary Table 2). Four variants, related to polyT stretches, were identified in a significant percentage of forward (88, 76, 74 and 75%) and reverse (94, 40, 40 and 14%) sequences but were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 2). Other five variants with lower percentages located in polyT stretches were not confirmed by the Sanger method (Figure 2; Supplementary Table 2). Unbalanced results with one sequence strand detected in a significant percentage (45–96%) but the other strand in a very low percentage (0–5%) were also not confirmed by Sanger sequencing (Figure 2; Supplementary Table 2). Variants found in a percentage of both strands below 25% were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 2).
Patient 3 (no. 3017)
A total of 16 745 301 bp represented by 70 358 reads (51.86%) with a mean read length of 240 bp passed the Genome Sequencer software's quality control system. The remaining percentage did not pass quality filters for short read length (37.05%) or incomplete extension and mixed reads (11.08%). A total of 117 variants were detected by the GS Junior system (Figure 2; Supplementary Table 3). Among them, 19 variants were confirmed by Sanger sequencing (Figure 2; Table 1). All of these variants (15 exonic and 4 intronic) are known polymorphisms except for two in the COL4A4 gene (NM_000092): 4749_4751delGTC (p.Gln1583_Ser1584SdelInsHis) and p.Arg1682Trp (c.5044C>T; Figures 2 and 4; Table 1).12, 24, 25, 26, 27, 28, 29 The first sequence variant was previously identified by DHPLC analysis and Sanger sequencing (Renieri A, unpublished data). This variant was inherited from the father (Figure 1). The second sequence variant, confirmed by Sanger sequencing, was not previously detected by DHPLC analysis and has been never reported as polymorphism (Figure 4; Table 1). We determined that the variation was inherited from the mother and was absent in the healthy sister and grandmother (Figure 1). The variation results in the substitution of the charged polar arginine with an aromatic nonpolar tryptophan in the noncollagenous domain of the protein, adjacent to a cysteine that forms disulfide bonds. Furthermore, arginine at position 1682 resulted highly conserved in different species (Supplementary Figure 2; http://genome.ucsc.edu/). However, PolyPhen (http://genetics.bwh.harvard.edu/pph) analysis, used to evaluate the likelihood that the observed variation alters the function of the protein, predicted the missense change to be probably benign.
Three out of 19 variants were present in 99 to 100% of the sequences and Sanger sequencing confirmed that they were present in homozygous/hemizygous state (Supplementary Table 3). A total of 14 variants were present in 57 to 20% of sequences (Supplementary Table 3). Sanger sequencing demonstrated that these variants were present in heterozygous state (Supplementary Table 3). One variant (IVS4-41T>G), identified in 91% of forward sequences and 26% of reverse sequences, was demonstrated to be in heterozygous state by Sanger Sequencing (Supplementary Table 3). One variant, related to a polyT stretch, was identified in a significant percentage of reverse (82%) sequences and in none of the forward sequences (Figure 2; Supplementary Table 3). It was not confirmed by Sanger sequencing and was considered as technical artifact. Unbalanced results with one sequence strand detected in a significant percentage (27–63%) but the other strand in a low percentage (0–2%) were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 3). Variants found in a percentage of both strands below 21% were not confirmed by Sanger sequencing (Figure 2; Supplementary Table 3).
Discussion
For the past 30 years, the Sanger method has been the dominant approach for DNA sequencing. The commercial launch of the first massively parallel pyrosequencing platform in 2005 ushered in the new era of high-throughput genomic analysis now referred to as NGS.30 Next-generation high-throughput DNA sequencing techniques have opened up fascinating new opportunities in biomedicine.31 For human genetics, there is an increasing need to analyze multiple genes that, when mutated, lead to overlapping physical findings and clinical phenotypes. In this study, for the first time, we applied GS Junior Sequencing (Roche 454) to simultaneously analyse three genes implicated in ATS: COL4A5, COL4A4 and COL4A3. The application of this technology allowed us to identify the second sequence variant in two Alport patients (Patient 2 and 3) and to revalue the diagnosis in a third patient (Patient 1).
Previous diagnoses of ATS have relied on DHPLC and/or Sanger sequencing.13, 18, 20, 32, 33, 34, 35 This diagnosis protocol is time-consuming and incurs relatively high costs. These pitfalls are because of the absence of hot spots in the genes in question and to the high number of exons per gene: 51 (COL4A5), 48 (COL4A4) and 52 (COL4A3). Usually, in Italy, 5–6 months are necessary to complete diagnosis, especially in cases with less informative pedigrees. Our data indicate that using 454 GS Junior sequencing, the diagnosis of ATS can be completed in 6 working days. Using the 454 technology also reduces costs. Conventional analysis of the three ATS genes in one patient can cost as much as ∼5000 Euros, whereas 454 technology can reduce this cost to ∼3000 Euros per patient. Further reduction in costs can be achieved by adding short nucleotide adapters (multiplex identifier (MID) sequences) as ‘barcodes’ between the tail and the specific sequence (Supplementary Table 4) and running multiple samples in the same experiment. By using two different MID sequences it is possible to analyze as many as four patients in a single experiment.
The NGS method allowed us to identify two not previously detected variations: p.Ser1147Phe in the COL4A3 gene (patient 2) and p.Arg1682Trp in the COL4A4 gene (patient 3; Supplementary Tables 2 and 3; Figures 3 and 4). The first one causes the substitution of the small uncharged polar serine with the aromatic nonpolar phenylalanine in the collagenous domain of the protein and, therefore, is expected to produce abnormal chains that can be incorporated into abnormal tropocollagen monomers. The other mutation, p.Arg1682Trp, results in the substitution of the charged polar arginine with an aromatic nonpolar tryptophan in the noncollagenous domain of the protein, adjacent to a cysteine that forms disulfide bonds essential for the stabilization of the protein structure. Although PolyPhen analysis predicted both amino acid changes to be likely benign, segregation studies and amino acids conservation data are in favour of a pathogenic role of the substitutions (Supplementary Figures 1 and 2). However, these conflicting results make more complex to definitively assess the pathogenicity of the variants and this represents an important problem for diagnostic laboratories.
Clinically, Patient 2 and Patient 3 had a confident diagnosis of ATS, whereas Patient 1 presented an atypical phenotype. This patient, now aged 19 years, exhibited isolated persistent proteinuria associated with normal renal function. He showed only one episode of gross hematuria at the age of 18 years. Ultrastructural analysis of renal biopsy showed thin glomerular basement membrane nephropathy. In this patient, GS Junior Sequencing did not identify any pathogenic mutation but only benign polymorphisms (Table 1). This result is consistent with the clinical picture of the patient and led us to reconsider the diagnosis of ATS and to revaluate the phenotype.
In this pilot study, we used Sanger sequencing as the gold standard method to evaluate the number of false-positive results and to determine indicative cut-off values to potentially use in a diagnostic setting (Figure 2; Supplementary Tables 1–3). Sanger sequencing revealed that variations detected in a percentage <25% of forward and reverse sequence reads were all false positives of 454 technology (Figure 2; Supplementary Tables 1–3). These sequence changes represent a high percentage of ‘not confirmed variations’ (87% in Patient 1, 82.5% in Patient 2 and 89% in Patient 3) and, therefore, this might be a useful signal for flagging possible false-positive results (Figure 2).
Furthermore, Sanger sequencing indicated that a small fraction of highly unbalanced data with one variation detected in a significant percentage (26–96%) of one sequence strand but in a very low percentage (0–5%) of the other strand should be considered as technical artifacts (Figure 2; Supplementary Tables 1–3). However, in Patient 1, Sanger sequencing confirmed an intronic variant (IVS46-8T>C in COL4A4) that was identified in 98% of forward sequences and 8% of reverse sequences, percentages very close to the range values indicating a technical artifact. In these cases, it is important to consider whether this variation is an already known SNP. IVS46-8T>C in COL4A4 has been previously reported as benign.12 It is not a candidate pathogenic change and its validation by Sanger sequencing is therefore not necessary in a diagnostic setting.
Finally, we found 18 variations located in polyT stretches that were not confirmed by Sanger sequencing (Figure 2; Supplementary Tables 1–3). Drawbacks of pyrosequencing include the fact that the signal intensity must be correlated with the number of bases incorporated, which proves problematic for the sequencing of homonucleotide regions that are greater than six bases in length. Because these repeated regions can represent hotspots for disease causing mutations, accurate detection of insertions/deletions in these stretches is very important. Technical and/or bioinformatic improvements should, therefore, be provided for diagnostic application.
Because the GS system identifies a high number of genetic variations, the usage of this platform in diagnostic laboratories requires the establishment of an analytic workflow to select candidate pathogenic changes, which need further follow-up. On the basis of the present and previous studies, we suggest the following selection criteria: (i) variations not corresponding to a known SNP and (ii) variations predicted to be truncating (premature stop/frameshift/splicing disruption) or altering an amino acid, (iii) variations with at least 30 × coverage depth and (iv) variations detected in >24% of reads.36, 37, 38 Although this analytic process would miss rare mutations in regulatory regions, it would be expected to detect most clinically relevant mutations.
In conclusion, in this work we present the first successful application of the 454 GS Junior Sequencing platform to simultaneously analysing three genes involved in ATS: COL4A5, COL4A4 and COL4A3. Although improvements will be necessary in accuracy and ease of data analysis, our study demonstrates that the system can be used to perform a fast, sensitive and relatively low-cost screening of variations in ATS genes.
References
Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG : Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med 2003; 348: 2543–2556.
Alport AC : Hereditary familial congenital hemorragic nephritis. Brit Med J 1927; 1: 504–506.
Flinter F : Alport’s syndrome. J Med Genet 1997; 34: 326–330.
Kashtan CE, Michael AF : Alport syndrome. Kidney Int 1996; 50: 1445–1463.
Wing A, Brunner F : Twenty-three years of dialysis and transplantation in Europe: experiences of the EDTA Registry. Am J Kidney Dis 1989; 14: 341–346.
Jais JP, Knebelmann B, Giatras I et al: X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 2000; 11: 649–657.
Pirson Y : Making the diagnosis of Alport’s syndrome. Kidney Int 1999; 56: 760–775.
Myers JC, Jones TA, Pohjolainen ER et al: Molecular cloning of a5(IV) collagen and assignment of the gene to the region of the X chromosome containing the Alport syndrome locus. Am J Hum Genet 1990; 46: 1024.
Flinter FA, Cameron JS, Chantler C, Houston I, Bobrow M : Genetics of classic Alport’s syndrome. Lancet 1988; ii: 1005–1007.
Kashtan CE : Alport syndrome and the X chromosome: implications of a diagnosis of Alport syndrome in females. Nephrol Dial Transplant 2007; 22: 1499–1505.
Jais JP, Knebelmann B, Giatras I et al: X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a ‘European Community Alport Syndrome Concerted Action’ Study. J Am Soc Nephrol 2003; 14: 2603–2610.
Longo I, Porcedda P, Mari F et al: COL4A3/A4 mutation: from benign familial hematuria to autosomal dominant or recessive Alport syndrome. Kidney Int 2002; 61: 1947–1956.
Marcocci E, Uliana V, Bruttini M et al: Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrol Dial Transplant 2009; 24: 1464–1471.
Smeets HJ, Lemmik HH, Van Den Heuvel LP : Molecular and immunological studies in X-linked and autosomal recessive in Alport syndrome. Am J Hum Genet 1993; 53: 1230.
Mochizuki T, Lemmink HH, Maryiama M et al: Identification of mutations in the a3(IV) and a4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994; 8: 77–82.
Knebelmann B, Benessy F, Buemi M, Grunfeld JP, Gubler MC, Antignac C : Autosomal recessive (AR) inheritance in Alport syndrome (AS). J Am Soc Nephrol 1993; 4: 263.
Jefferson JA, Lemmink HH, Hughes AE et al: Autosomal dominant Alport Syndrome linked to the type IV collagen a3 and a4 genes (COL4A3 and COL4A4). Nephrol Dial Transplant 1997; 12: 1595–1599.
van der Loop FT, Heidet L, Timmer ED et al: Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int 2000; 58: 1870–1875.
Ciccarese M, Casu D, Ki Wong F et al: Identification of a new mutation in the alpha4(IV) collagen gene in a family with autosomal dominant Alport syndrome and hypercholesterolaemia. Nephrol Dial Transplant 2001; 16: 2008–2012.
Pescucci C, Mari F, Longo I et al: Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int 2004; 65: 1598–1603.
Nyren P, Pettersson B, Uhlen M : Solid phase DNA minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Anal Biochem 1993; 208: 171–175.
Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P : Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996; 242: 84–89.
Ronaghi M, Uhlen M, Nyren P : A sequencing method based on real-time pyrophosphate. Science 1998; 281: 363, 365.
Barker DF, Denison JC, Atkin CL, Gregory MC : Efficient detection of Alport syndrome COL4A5 mutations with multiplex genomic PCR-SSCP. Am J Med Genet 2001; 98: 148–160.
Tazon Vega B, Badenas C, Ars E et al: Autosomal recessive Alport’s syndrome and benign familial hematuria are collagen type IV diseases. Am J Kidney Dis 2003; 42: 952–959.
Badenas C, Praga M, Tazon B et al: Mutations in theCOL4A4 and COL4A3 genes cause familial benign hematuria. J Am Soc Nephrol 2002; 13: 1248–1254.
Boye E, Mollet G, Forestier L et al: Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport Syndrome. Am J Hum Genet 1998; 63: 1329–1340.
Lemmink HH, Mochizuki T, van der Heuvel LP et al: Mutations in the type IV collagen a3(COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 1994; 3: 1269–1273.
Heidet L, Arrondel C, Forestier L et al: Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport Syndrome. J Am Soc Nephrol 2001; 12: 97–106.
Voelkerding KV, Dames S, Durtschi JD : Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn 2010; 12: 539–551.
Ansorge WJ : Next-generation DNA sequencing techniques. N Biotechnol 2009; 25: 195–203.
Nagel M, Nagorka S, Gross O : Novel COL4A5, COL4A4, and COL4A3 mutations in Alport syndrome. Hum Mutat 2005; 26: 60.
Longo I, Scala E, Mari F et al: Autosomal recessive Alport syndrome: an in-depth clinical and molecular analysis of five families. Nephrol Dial Transplant 2006; 21: 665–671.
Hou P, Chen Y, Ding J, Li G, Zhang H : A novel mutation of COL4A3 presents a different contribution to Alport syndrome and thin basement membrane nephropathy. Am J Nephrol 2007; 27: 538–544.
Tan R, Colville D, Wang YY, Rigby L, Savige J : Alport retinopathy results from ‘Severe’ COL4A5 mutations and predicts early renal failure. Clin J Am Soc Nephrol 2010; 5: 34–38.
Raca G, Jackson C, Warman B, Bair T, Schimmenti LA : Next generation sequencing in research and diagnostics of ocular birth defects. Mol Genet Metab 2010; 100: 184–192.
Smith DR, Quinlan AR, Peckham HE et al: Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res 2008; 18: 1638–1642.
Chou LS, Liu CS, Boese B, Zhang X, Mao R : DNA sequence capture and enrichment by microarray followed by next-generation sequencing for targeted resequencing: neurofibromatosis type 1 gene as a model. Clin Chem 2010; 56: 62–72.
Acknowledgements
We first thank ATS patients and their families. We also thank Dr Michele Iacono for his technical support. This work was supported by Regione Toscana project (Progetto Regionale malattie rare) to AR and by a donation to AR ideally dedicated to ‘Graziano and Marco Laurini’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Artuso, R., Fallerini, C., Dosa, L. et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet 20, 50–57 (2012). https://doi.org/10.1038/ejhg.2011.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.164
Keywords
This article is cited by
-
Variants in genes coding for collagen type IV α-chains are frequent causes of persistent, isolated hematuria during childhood
Pediatric Nephrology (2023)
-
New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells
European Journal of Human Genetics (2020)
-
Expert consensus guidelines for the genetic diagnosis of Alport syndrome
Pediatric Nephrology (2019)
-
A protocol for the identification and validation of novel genetic causes of kidney disease
BMC Nephrology (2015)
-
Clinical utility gene card for: Alport syndrome – update 2014
European Journal of Human Genetics (2015)
