
Abstract
We report on three hydropic fetuses of 17, 22 and 25 gestational weeks from three distinct families presenting with Desbuquois dysplasia type 1. All fetuses showed brachymelia and characteristic dysmorphic features. X-ray studies revealed δ-shaped extraphalangeal bones and disease-specific prominence of the lesser trochanter, varying in severity with fetal age. Early lethal manifestation of the disorder was reflected in lung hypoplasia and in early death of similarly affected siblings in cases 1 and 2. All families were German Caucasians by descent. Sequence analysis of the CANT1 gene revealed two frameshift mutations, c.228_229insC and c.277_278delCT, in homozygous and compound heterozygous configuration, respectively, and a homozygously novel missense mutation, c.336C>A (p.D112E), located within a highly conserved region of exon 2. Haplotype analyses by high-resolution single-nucleotide polymorphism array showed that the haplotype associated with c.228_229insC may be traced to a single founder in the German population.
Similar content being viewed by others

Introduction
Autosomal recessive Desbuquois dysplasia (DBQD, OMIM 251 450) is a skeletal disorder defined by pre- and postnatal growth failure, short limbs, spondylometaphyseal abnormalities, osteopenia, advanced carpotarsal ossification, and ligamentous joint laxity. It is associated with characteristic dysmorphic features, including a flat round face, midface hypoplasia, short nose, microstomia, and microretrognathia, often resulting in a Pierre Robin anomaly. Characteristic radiologic features are a small δ-shaped extraphalangeal bone, distal to the second metacarpal, leading to radial deviation of the index fingers, metaphyseal flaring with prominent lesser trochanter, causing a monkey wrench appearance of the proximal femora, horizontal acetabular roofs with dislocation of femoral heads, narrow thorax, and coronal or sagittal clefting of the vertebrae.1, 2, 3, 4, 5, 6 There are cases with mild clinical features that show no prenatal growth failure, normal hands, and only minor facial dysmorphism and joint problems. Thus, the disorder has been divided into two distinct groups with normal hands (type 2), and with abnormal hands (type 1).7, 8 The latter condition displays a high lethality rate of >33% due to respiratory failure.9 Sex distribution in 20 documented cases of DBQD type 1 is 3:2 (male:female).3 Recently, a new clinical subtype of DBQD has been described.10
The disease gene was localized to 17q25.3.11 In 2009, mutations in the calcium-activated nucleotidase (CANT1) gene were identified to be responsible for DBQD type 1 in 10 affected patients from nine, mostly non-European families. Early (perinatal) lethality in four of these patients was associated with nonsense mutations.12 Very recently, mutations in the CANT1 gene have also been found associated with the DBQD type 2, as well as with the so called ‘Kim variant’ of DBQD.13
Case descriptions
Case 1
The first son of a 26-year-old woman died a few days after delivery from respiratory failure. He was affected by a skeletal dysplasia that, due to the presence of an atrioseptal defect, triphalangeal thumbs, and supernumerary ossicles in both hands, was classified as Ellis–van Creveld syndrome. Ten years later, recurrence of Ellis–van Creveld syndrome was diagnosed in the second pregnancy by prenatal ultrasound. The pregnancy was terminated at 25 gestational weeks (g.w.). The healthy parents were consanguineous by an uncle–niece relationship and of German Caucasians descent (Supplementary Figure s1a)
Case 2
A male fetus resulted from the third pregnancy of non-consanguineous, healthy, 30-year-old parents of German Caucasians descent. No information was available about common ancestors. Their first-born girl had been affected by an unclassified chondrodysplasia, and had died at the age of 5 months from respiratory problems. The second-born son, as also a fourth-born daughter are healthy. Prenatal ultrasound during the third pregnancy revealed a recurrence of chondrodysplasia. The pregnancy was terminated at 21 g.w. (Supplementary Figure s1b).
Case 3
Prenatal ultrasound during the third pregnancy of a healthy 29-year-old nullipara, with two previous early abortions, revealed fetal hydrops with hygroma colli and short long bones. The pregnancy was terminated at 17+1 g.w. Both non-consanguineous parents were German Caucasians by descent and no information was available about common ancestors (Supplementary Figure s1c).
Autopsy:
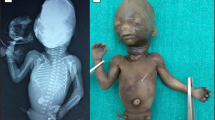
The three hydropic male fetuses weighed 600/360/111 g, were 17.5/15/10 cm crown-rump length, and 23.5/22.5/13.8 cm crown-heel length. They displayed almost identical characteristics, namely, brachymelic dwarfism, short neck and trunk, distended abdomen, and a large cranium with high forehead (Figures 1a and b). The faces were round and flat with short midfaces, very short, flat and broad noses with anteverted nares, microstomia, and microretrognathia, associated with a Pierre Robin anomaly in cases 1 and 2. The eyes were upslanting with proptosis and epicanthic folds, and the ears were dorsally rotated and overfolded. In addition, case 3 had cystic hygroma of the neck (Figure 1c). The hands of each case showed radial deviation and clinodactyly of index fingers, and low inserting broad thumbs with dysplastic nails. The feet were club feet, with low inserting and short big toes. X-ray examination revealed mild shortening and bowing of long bones with slightly flared metaphyses, and a protuberant lesser trochanter, in case 1, giving a monkey wrench appearance of the proximal femora. There was pronounced shortening of the first metatarsals and of the first and second metacarpals, duplication or splitting of the distal phalanx of the thumbs, and a small laterally displaced extra phalangeal bone at the base of both index fingers (Figures 2a–c). The femoral heads appeared dislocated, with narrow horizontal acetabular roofs (Figures 3a–c). Other joints were fixed by the hydropic surrounding tissues, prohibiting an assessment of joint laxity in the three fetuses. The thorax was narrow and bell shaped, and the ribs were thin. The vertebral bodies showed sagittal clefts in the lumbar and thoracic regions of the spine in cases 1 and 2, and more severe ossification defects of the spine in case 3 (Supplementary Figure s2). Histological examination of the long bones revealed crowding of enlarged resting chondrocytes, and short, irregular columns with ovoid groupings of chondrocytes, as described by Shohat et al.6 In addition, there were distinctly hypoplastic lungs, laryngeal stenosis, and dilatation of ureters and renal pelvices in case 1, ASD in the affected sib of case 1, horseshoe kidneys in case 2, laryngo-tracheomalacia in the affected sib of case 2, and subaortic septal hypertrophy in case 3. Considering the characteristic findings and the affected sibs, (recurrent) DBQD type 1 was diagnosed in all three hydropic fetuses (Supplementary Table s1).

Fetal aspect of case 1, 2, and 3 showing short limbs and trunk, hydrops, large cranium with flat round face, severe microretrognathia, and brachydaktyly, with short broad thumbs and radial deviation of index fingers. (a) Case 1 – male, 25 g.w.; (b) case 2 – male, 21 g.w.; (c) case 3 – male, 17 g.w.

X-ray of hands demonstrating short phalanges, short first and second metacarpals, duplicated or splitted distal phalanges of the thumbs, and radial deviation of index fingers due to an extraphalangeal bone at the base of the proximal phalanges (arrows). (a) Left hand of case 1, (b) right hand of case 2, (c) right hand of case 3.

X-ray of femora and hips displaying narrow and horizontal acetabular roofs, mild shortening and bowing of femora, slight flaring of femoral metaphyses, and protuberance of the lesser trochanter, giving a distinct monkey wrench appearance only in case 1 (a, arrows). (b) Case 2 and (c) case 3.
Molecular studies
DNA was extracted from deep-frozen fetal tissues in cases 2 and 3, using standard procedures. For the only available paraffin embedded tissues in case 1 (lung and kidney), the DNA was isolated using the EZ1 DNA Tissue Kit from Qiagen (Hilden, Germany). The four exons of the CANT1 gene (transcript ENST00000302345) and adjacent intronic sequences were PCR amplified with newly designed primers (Supplementary Table s2), and sequenced using a fluorescent automated sequencer (3130xl Genetic Analyzer; Applied Biosystems, Foster City, CA, USA). The fetus in case 1 was homozygous for a novel missense mutation c.336C>A in exon 2 of the CANT1 gene, predicted to result in the replacement of aspartate with glutamate at residue 112 (p.D112E) (Supplementary Figures s3a and b). Case 2 displayed compound heterozygosity for two frameshift mutations in exon 2, c.277_278delCT (p.L93VfsX89), which is novel, and c.228_229insC (p.W77LfsX13) (Supplementary Figures s4 and s5). The latter mutation c.228_229insC was also found to be homozygous in case 3. Analysis of parental DNA in cases 2 and 3 confirmed Mendelian inheritance of the mutations. Parental DNA in case 1 was unavailable, but homozygosity was in line with the reported consanguinity of the parents. The presence of the mutations c.336C>A and c.228_229insC has been also confirmed by RFLPs analysis (data not shown). In case of the c.277_278delCT mutation, no suitable restriction site was available for an RFLP analysis. In order to determine whether the mutation c.228_229insC can be traced to a single founder, we carried out haplotype analyses on genomic DNA of the parents and fetuses of case 2 and 3, using a high-resolution single-nucleotide polymorphism (SNP) array analysis (Illumina Omni1-quad Beadchips, according to the Infinum HD Assay Protocol, Illumina, San Diego, CA, USA). Case 3 carried a rare haplotype in homozygous form for an 865 kb region on chromosome 17, which includes the CANT1 gene. Haplotype retracing confirmed that the (maternally inherited) mutation c.228_229insC in case 2 was associated with the same SNP haplotype. This joint haplotype was limited by SNPs rs9910295 and rs2703549 on chromosome 17, an area that represents a known haplotype block. SNP alleles in the narrow, stable haplotype block containing the CANT1 gene did not correspond to one of the predicted HapMap haplotypes,14 but in 6/8 positions resembled a HapMap haplotype with an allele frequency of 0.132. Our examination of published SNP frequency data and our own data confirmed an allele frequency of the haplotype associated with c.228_229insC of at most 12%. The data thus indicate that mutation c.228_229insC may be traced to a single founder in the German population. Molecular analysis of 200 chromososmes in a panel of 200-unrelated German Caucasian healthy controls excluded the presence of both c.336C>A and c.228_229insC mutations.
Discussion
All three hydropic fetuses were affected by DBQD type 1, as confirmed by mutations in the CANT1 gene. Independent of fetal age, they all displayed short-limb dwarfism, radial deviation of index fingers due to a small δ-shaped extraphalangeal bone, duplicated, partly synostotic (split) distal phalangeal bones of the thumbs, horizontal acetabular roofs, underossification of vertebrae, and characteristic craniofacial features, associated with a Pierre Robin anomaly in cases 1 and 2 (Figures 1a and b, and 2a–c). However, the syndrome-specific ‘monkey wrench appearance’ of the proximal femora, caused by a prominent lesser trochanter, was convincingly demonstrated only in case 1, at 25 g.w. This abnormality was less evident in case 2, at 21 g.w., and not apparent in case 3, at 17 g.w., and thus represents an evolutive feature (Figures 3a–c).
As in other skeletal dysplasias, fetal hydrops implies a severe, lethal variant of the disorder. It has been mentioned in two sib fetuses of 12 and 19 g.w. that had shown similarities to DBQD. However, molecular studies for confirmation of the suspected diagnosis had not been performed.15 Hydrops was most marked in case 3 with cystic hygroma of the neck that has not been described before. In the absence of a previous sib with skeletal dysplasia, this hydrops triggered a more thorough fetal ultrasound examination. In cases 1 and 2, diagnosis of DBQD allowed correction of the diagnoses in the previously born sibs. Failure to classify a skeletal dysplasia as DBQD type 1 in a prenatal ultrasound is well documented in the literature. As discussed by Baynam et al,15 an ‘upsilon sign’ illustrating the widely splayed digits 2 and 3, as caused by the extraphalangeal bones, may be used as a specific clue in prenatal ultrasound, and a molecular analysis may provide diagnostic certainty. In addition to the characteristic features of DBQD, our cases presented with an atrial septal defect in the affected sib of case 1, with subaortic septal hypertrophy in case 3, and with renal anomalies, such as uretero-pelvic dilatation, horseshoe kidney, and single renals cysts in cases 1, 2, and 3. While structural heart defects had been described before,2, 12 renal anomalies have not yet been observed in DBQD. According to the Electronic Northern Expression, as well as to the Microarray Expression for CANT1, the gene is expressed in both heart and kidney (http://bioinfo2.weizmann.ac.il/cgi-bin/genenote/home_page.pl), and may have a role during the embryonic development of both organs.
DBQD must be differentiated clinically from Catel–Manzke, and from Larsen syndromes. An intermediate phenotype of DBQD, diastrophic dysplasia, and recessive multiple epiphyseal dysplasia, including the monkey wrench appearance of the proximal femora, have been identified with compound heterozygote DTDST gene mutations.16, 17
We identified three novel CANT1 gene mutations in our patients, all located within exon 2 of the gene. Two were frameshift mutations with predicted stop codons after 89 and 13 missense amino acid residues, respectively, and were expected to cause complete loss of protein function, probably through nonsense-mediated RNA decay as disease-causing mechanism. The two frameshift mutations were found in compound heterozygous form in case 2, and the c.228_229insC mutation in homozygous form in the severely hydropic fetus of case 3. The third mutation, found homozygously in the case with parental consanguinity, involves Asp112, an amino acid that is highly conserved up to maize (Supplementary Figure s3c). Yang and Kirley18 demonstrated that hSCAN-1, a protein of the apyrase family, binds poorly to the Cibacron blue nucleotide-analog affinity matrix in the presence of calcium if aspartic acid at position 84 is mutated (p.D84A); Asp84 of hSCAN-1 corresponds to Asp114 of the CANT1 protein. The homologous amino acids Asp112 and Asp114 are located in a highly conserved ‘nucleotide conserved region’ (NCR1 b) of the apyrase gene family. This domain may have a crucial role in the binding of nucleotides or may be important for calcium-dependent conformational changes of the protein. Hamasaki et al19 show that the D112 is indeed a key residue for nucleotide binding by mutagenic experiments. This missense mutation was also predicted to be probably damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00) by an in silico analysis, using the PolyPhen-2 software (v2.0.23r349, http://genetics.bwh.harvard.edu/pph2/).20 The mutation p.D112E is thus very likely to cause a complete loss-of-protein function in line with early lethality in our case 1.
Fifteen out of the 17 previously described families with DBQD were from different non-European ethnicities; one was from France, and a second from an Australia/Caucasian family.12, 13, 21 Sixteen different mutations were identified in these families (Table 1). Here we report the first mutations found in patients of German descent. Interestingly, three out of five independent DBQD alleles carried the same novel frameshift insertion, c.228_229insC. Haplotype analysis confirmed that this mutation most likely traces back to a single founder. It is possible that this mutation represents a relatively common mutation in the German population; prenatal lethality associated with this mutation may explain why this mutation has not been reported before in the German population. Notably, Furuichi et al13 reported this mutation in an Australian Caucasian patient. According to our results, it may be plausible that this patient has German ancestors. The identification in this patient of a common haplotype would reinforce our findings of a single founder for the c.228_229insC mutation. We are not able to reliably determine the age of this mutation, as the common haplotype encompasses a haplotype block, and there are only three independent alleles. The observations that three out of five independent chromosomes from our German patients carried this mutation may indicate that it is an old mutation, which may be expected also in other families.
The present report, in conjunction with the previously reported cases, confirms that complete functional loss of the CANT1 protein due to homozygous or compound heterozygous early-frameshift mutations, or missense mutations in critically functional regions of the protein is responsible for the early lethal form of DBQD. By contrast, missense mutations causing partial loss of protein function (eg, the recurrent p.R300C and R300H mutations in exon 4) are associated with the classical form of DBQD. It is also possible that some missense mutations may cause intermediate or abortive forms of DBQD. Furthermore, we unveiled a founder mutation (c.228_229insC) as demonstrated by the haplotype analysis in the German population.
Of our three fetal DBQD cases and the two affected sibs, four were male. This is consistent with previous reports that suggest a male preponderance of 2:3 F/M in DBQD type 1,3 and of 1:4 (4:17) F/M in ‘early lethal DBQD’.1, 3, 6, 9, 12, 21, 22 A good explanation for a male preponderance is still lacking; one possibility would be a better prenatal survival of male fetuses.15
References
Beemer FA, Kramer PP, van der Harten HJ, Gerards LJ : A new syndrome of dwarfism, neonatal death, narrow chest, spondylometaphyseal abnormalities, and advanced bone age. Am J Med Genet 1985; 20: 555–558.
Desbuquois G, Grenier B, Michel J, Rossignol C : Nanisme chondrodystrophique avec ossification anarchic et polymalformations chez deux soeurs. Arch Franc Pediatr 1966; 23: 573–587.
Gillessen-Kaesbach G, Meinecke P, Ausems MG et al: Desbuquois syndrome: three further cases and review of the literature. Clin Dysmorphol 1995; 4: 136–144.
Le Merrer M, Young ID, Stanescu V, Maroteaux P : Desbuquois syndrome. Eur J Pediatr 1991; 150: 793–796.
Meinecke P, Spranger J, Schaefer E, Maroteaux P : Micromelic dwarfism with vertebral and metaphyseal abnormalities and advanced carpotarsal ossification: another observation. Am J Med Genet 1989; 32: 432–434.
Shohat M, Lachman R, Gruber HE et al: Desbuquois syndrome: clinical, radiographic, and morphologic characterization. Am J Med Genet 1994; 52: 9–18.
Faivre L, Cormier-Daire V, Eliott AM et al: Desbuquois dysplasia, a reevaluation with abnormal and ‘normal’ hands: radiographic manifestations. Am J Med Genet A 2004; 124A: 48–53.
Nishimura G, Hong HS, Kawame H, Sato S, Cai G, Ozono K : A mild variant of Desbuquois dysplasia. Eur J Pediatr 1999; 158: 479–483.
Hall BD : Lethality in Desbuquois dysplasia: three new cases. Pediatr Radiol 2001; 31: 43–47.
Kim OH, Nishimura G, Song HR et al: A variant of Desbuquois dysplasia characterized by advanced carpal bone age, short metacarpals, and elongated phalanges: report of seven cases. Am J Med Genet A 2010; 152A: 875–885.
Faivre L, Le Merrer M, Al-Gazali LI et al: Homozygosity mapping of a Desbuquois dysplasia locus to chromosome 17q25.3. J Med Genet 2003; 40: 282–284.
Huber C, Oules B, Bertoli M et al: Identification of CANT1 mutations in Desbuquois dysplasia. Am J Hum Genet 2009; 85: 706–710.
Furuichi T, Dai J, Cho TJ et al: CANT1 mutation is also responsible for Desbuquois dysplasia, type 2 and Kim variant. J Med Genet 2010; 48: 32–37.
Frazer KA, Ballinger DG, Cox DR et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Baynam G, Kiraly-Borri C, Goldblatt J, Dickinson JE, Jevon GP, Overkov A : A recurrence of a hydrop lethal skeletal dysplasia showing similarity to Desbuquois dysplasia and a proposed new sign: the Upsilon sign. Am J Med Genet A 2010; 152A: 966–969.
Miyake A, Nishimura G, Futami T et al: A compound heterozygote of novel and recurrent DTDST mutations results in a novel intermediate phenotype of Desbuquois dysplasia, diastrophic dysplasia, and recessive form of multiple epiphyseal dysplasia. J Hum Genet 2008; 53: 764–768.
Panzer KM, Lachman R, Modaff P, Pauli RM : A phenotype intermediate between Desbuquois dysplasia and diastrophic dysplasia secondary to mutations in DTDST. Am J Med Genet A 2008; 146A: 2920–2924.
Yang M, Kirley TL : Site-directed mutagenesis of human soluble calcium-activated nucleotidase 1 (hSCAN-1): identification of residues essential for enzyme activity and the Ca(2+)-induced conformational change. Biochemistry 2004; 43: 9185–9194.
Hamasaki R, Kato H, Terayama Y, Iwata H, Valenzuela JG : Functional characterization of a salivary apyrase from the sand fly, Phlebotomus duboscqi, a vector of Leishmania major. J Insect Physiol 2009; 55: 1044–1049.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Faden M, Al-Zahrani F, Arafah D, Alkuraya FS : Mutation of CANT1 causes Desbuquois dysplasia. Am J Med Genet A 2010; 152A: 1157–1160.
al-Gazeli LI, Aziz SA, Bakalinova D : Desbuquois syndrome in an Arab Bedouin family. Clin Genet 1996; 50: 255–259.
Acknowledgements
We are grateful to Dr Ute Moog, Institute of Human Genetics, University Heidelberg, for additional information concerning case 1 and to Dr Jürgen W. Spranger for clinical advice in case 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Laccone, F., Schoner, K., Krabichler, B. et al. Desbuquois dysplasia type I and fetal hydrops due to novel mutations in the CANT1 gene. Eur J Hum Genet 19, 1133–1137 (2011). https://doi.org/10.1038/ejhg.2011.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.101
Keywords
This article is cited by
-
Antenatal Phenotype of Desbuquois Dysplasia
Indian Journal of Pediatrics (2023)
-
Prenatal exome sequencing and chromosomal microarray analysis in fetal structural anomalies in a highly consanguineous population reveals a propensity of ciliopathy genes causing multisystem phenotypes
Human Genetics (2022)
-
Prenatal diagnosis of fetal skeletal dysplasia using 3-dimensional computed tomography: a prospective study
BMC Musculoskeletal Disorders (2020)
-
Cloning, expression and enzyme activity delineation of two novel CANT1 mutations: the disappearance of dimerization may indicate the change of protein conformation and even function
Orphanet Journal of Rare Diseases (2020)
-
Posterior vertebral column resection for rigid proximal thoracic kyphoscoliosis with broken growing rods in a patient with Desbuquois dysplasia
Spine Deformity (2020)
