
Abstract
Genome-wide high-resolution array analysis is rapidly becoming a reliable method of diagnostic investigation in individuals with mental retardation and congenital anomalies, leading to the identification of several novel microdeletion and microduplication syndromes. We have identified seven individuals with duplication on chromosome 14q11.2q13.1, who exhibited idiopathic developmental delay and cognitive impairment, severe speech delay, and developmental epilepsy. Among these cases, the minimal common duplicated region on chromosome 14q11.2q13.1 includes only three genes, FOXG1, C14orf23, and PRKD1. We propose that increased dosage of Forkhead Box G1 (FOXG1) is the best candidate to explain the abnormal neurodevelopmental phenotypes observed in our patients. Deletions and inactivating mutations of FOXG1 have been associated with a Rett-like syndrome characterized by hypotonia, irritability, developmental delay, hand stereotypies, and deceleration of head growth. FOXG1, encoding a brain-specific transcription factor, has an important role in the developing brain. In fact, in vivo studies in chicken brain demonstrated that overexpression of FOXG1 results in thickening of the neuroepithelium and outgrowth of the telencephalon and mesencephalum, secondary to a reduction in neuroepithelial cell apoptosis.
Similar content being viewed by others


Introduction
Deletions and inactivating mutations of the Forkhead Box G1 (FOXG1) gene on chromosome 14q12 have been associated with a Rett-like neurodevelopmental disorder characterized by developmental delay, hypotonia, irritability, repetitive hand stereotypies, and deceleration of head growth (OMIM 613454).1, 2, 3, 4, 5, 6, 7 The phenotypic similarities between classic Rett syndrome (OMIM 312750), which results from mutations or deletions in the methyl-CpG binding protein 2 (MECP2) gene, and the neurodevelopmental disorder caused by mutations or deletions in FOXG1 suggest that the disruption of one or more common neuronal developmental pathway(s) is responsible for these conditions. Nonetheless, important clinical differences exist between the two syndromes. For example, patients with FOXG1 mutations/deletions tend to exhibit abnormal development in early infancy, while also lacking the characteristic autonomic disturbances of Rett syndrome.
In male patients, duplications of MECP2 result in delayed milestones, infantile hypotonia, progressive spasticity, severe mental disability, absence of language, and increased susceptibility to recurrent infections (OMIM 300260).8 Interestingly, there is a significant phenotypic overlap between the neurological abnormalities seen in patients with increases or decreases in MECP2 dosage.9, 10 To date, duplication of the chromosomal region encompassing FOXG1 has been reported in only one patient with infantile spasms, severe intellectual impairment, and minor dysmorphisms.11
Here, we present the clinical and molecular findings in six patients with duplications of the 14q12 region containing the FOXG1 gene. A query of the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER; https://decipher.sanger.ac.uk)12 revealed a seventh patient (DECIPHER 248559) presenting with developmental delay and cognitive impairment and a 14q12 duplication involving the FOXG1 gene.
Materials and methods
DNA samples
Patients were referred for clinical array-CGH analysis to either Medical Genetics Laboratories at Baylor College of Medicine (BCM) in Houston, TX, USA (cases 1–3) or to the Cytogenetic Laboratory of the University of Pavia in Pavia, Italy (cases 4–6). Case 7 was obtained through a query of the DECIPHER database and is referred to as DECIPHER 248559.
Once informed consents, approved by the Institutional Review Board for Human Subject Research at the corresponding institutions, were obtained, DNA samples were collected from the probands and their family members.
DNA was extracted from whole blood using the Puregene DNA extraction kit (Gentra, Minneapolis, MN, USA) according to the manufacturer's instructions.
Array CGH
The 14q duplications in cases 1–3 were identified by screening the Medical Genetics Laboratories at BCM database of array CGH results for over 11 000 patients tested using oligonucleotide-based Chromosomal Microarray Analysis (CMA Versions 6, 7, and 8 OLIGO, Baylor College of Medicine, Houston, TX, USA).13, 14 Approximately 4700 of these patients were tested using the v6 OLIGO (44K array), 5145 patients using the v7 OLIGO (105K array), and 1800 using the v8 OLIGO (180K array). The rearrangements in patients 2 and 3 were further investigated using a high-resolution 244K Agilent Technologies (Santa Clara, CA, USA) and 2.1 M Roche NimbleGen arrays (Madison, WI, USA), respectively. Cases 4-6 were identified by the cytogenetics laboratory at the University of Pavia, from a search of the 3752 cases analyzed. Patient 5 was evaluated using array-CGH with a 44K array (Agilent Technologies). Patients 4 and 6 were first examined with the same 44K platform before running them on a 180K array (Agilent). All experiments were performed according to the manufacturer's instructions with some modifications.15 The slides were scanned into image files using a GenePix Model 4000B microarray scanner (Molecular Devices, Sunnyvale, CA, USA) or an Agilent G2565 laser scanner. Microarray image files of oligo arrays were quantified using Nimblescan v2.5 (Roche NimbleGen) or Feature Extraction version 9.0 (Agilent Technologies), and text file outputs from the quantitation analysis were imported to SignalMap v1.9 (NimbleGen Systems), CGH Analytics Software v4.0.85 (Agilent Technologies), or our in-house copy number analysis package, as described.14
SNP microarray
Patient 6 (DECIPHER 248559) was evaluated using a 250K GeneChip Human Mapping Array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's specifications.
Long-range PCR and DNA sequencing
Long-range PCR used to amplify the predicted junction fragments from the breakpoint regions in patient 3, which was performed as per the manufacturer's instructions (Takara Bio Inc., Otsu, Japan). PCR products were purified with the PCR Purification Kit (Qiagen, Valencia, CA, USA) and bidirectionally sequenced using the Sanger's dideoxynucleotide method (Lone Star, Houston, TX, USA).
Bioinformatics and in silico sequence analysis
Genomic sequences based on the oligonucleotide coordinates from the array CGH experiment were downloaded from the UCSC genome browser (Build 36, UCSC genome browser, March 2006). Interspersed repeat sequences were analyzed by RepeatMasker (http://www.repeatmasker.org). Regional assemblies were done using NCBI BLAST 2 and the Sequencher software (Gene Codes, Ann Arbor, MI, USA).
Patient reports
Patient 1
This 4-year-old boy was initially evaluated for a history of infantile spasms and global developmental delay. He was born with unilateral postaxial polydactyly, which was corrected surgically, and he underwent strabismus repair at 2 years of age. At the age of 6 months, he developed infantile spasms. The electroencephalogram (EEG) performed at that time documented hypsarrhythmia with electrodecrement. Therapy was initiated with adrenocorticotropic hormone, and a repeat EEG performed 1 month later showed improvement in the background rhythm, although there was a persistence of both right lateral sharp waves and right lateral diffuse slowing. He has been seizure-free on lamotrigine for 3 years and his most recent EEG at the age of 4 years showed an 8 Hz posterior dominant rhythm.
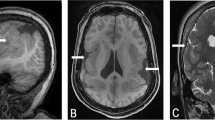
His developmental delays included not walking until the age of 3 years and an unstable gait with frequent falls at 4 years, with additional lack of speech, unexpected and inappropriate laughing, and not following commands. However, there was no history of regression. He also exhibited consistently disrupted sleep. On physical examination at 3 years and 5 months of age, he weighed 14.2 kg (25–50th centile), with a height of 100 cm (50–75th centile), and a head circumference of 49.1 cm (−3SD). He was non-dysmorphic (Figure 1a) and on neurological examination, it was discovered that he had nystagmus, walked with a wide base gait, and preferred walking on his toes. He also exhibited perseverative, stereotyped hand movements. A brain MRI at 6 months revealed mildly foreshortened frontal lobes, and an intact corpus callosum (Figure 2a and b).

Facial appearance of individuals with the duplication involving 14q11.2q13.1 (a: patient 1; b: patient 2; c: patient 3; d: patient 4; e: patient 5; f: patient 6; g: DECIPHER case 248559). Patients 2, 6, and DECIPHER 248559 have facial dysmorphisms. Patient 1, 4, and 5 are non-dysmorphic. Only mild dysmorphisms are seen in patient 3.

Brain MRI of patient 1 showed an intact anterior corpus callosum (arrow) (a) and mildly foreshortened frontal lobes (b). Brain MRI of patient 3 showed thin corpus callosum (arrow) (c), reduction in white matter volume, large lateral ventricles, and increased periventricular white matter signal (d).
Patient 2
This female patient was first evaluated at the age of 3 years for developmental delay and seizures. She was born at term of an uncomplicated gestation by spontaneous vaginal delivery to a 36-year-old mother. She began suffering from generalized tonic clonic seizures at the age of 6 months. The EEG at 3 years and 3 months showed epileptiform activity in the form of multifocal spiking and slow wave activity, as well as some bursts of generalized spiking and slow wave activity. The background activity and the occipital dominant rhythm were both slightly slow for her age. The seizures were well controlled beginning at the age of 2 years with zonisamide monotherapy. Her developmental delays included being unable to sit without support or crawl before she turned 1-year, as well as the inability to walk independently until 30 months of age. Additionally, her speech was significantly delayed and she had only one word at the time of evaluation. On physical examination at 3 years, her weight was 15.1 kg (75th percentile), her height 88 cm (5th percentile), and her head circumference 48 cm (25th percentile). She presented with dysmorphic facies with mid-face hypoplasia, a flat nasal bridge, mild hypotelorism, and short palpebral fissures (Figure 1b). She also had bilateral, malformed and cupped ears, bilateral ear pits, bilateral fifth finger clinodactyly, and right fifth finger brachydactyly. A brain MRI revealed only a mildly small posterior fossa and foreshortening of the corpus callosum.
Patient 3
At 6 months of age, this male patient was evaluated for infantile spasms and developmental delay. He was born after 41 weeks of gestation to a 37-year-old mother by cesarean section. The pregnancy was complicated by maternal varicella infection in the fifth month of pregnancy. A prenatal ultrasound revealed the presence of a choroid plexus cyst. At birth, the patient weighed 3317 g (25–50th centile) with a length of 52.1 cm (75–90th centile), and he had a right clubfoot. The patient developed infantile spasms at 3 months of age and subsequently began treatment with antiepileptic medications (ACTH, prednisone, and topiramate). An initial EEG confirmed the presence of epileptiform activity consistent with modified hypsarrhythmia. After antiepileptics, a follow-up EEG showed diffuse background slowing for his age, as well as the presence of focal delta slowing over the left temporal region with intermixed high-amplitude and irregular, spiked and slow wave discharges. These data indicated diffuse and focal cerebral dysfunction, and was more prominent over the left temporal region, and was also epileptogenic.
At 13 months, his developmental delays included the inability to sit up, occasional rolling over, absence of speech, and no reaching out or following. Both parents were healthy, but the mother reported a history of recurrent spontaneous miscarriages. At the age of 13 months, the patient's growth parameters were within the normal range, including a head circumference of 46 cm (25–50th centile). The patient's physical exam showed normal growth parameters and only mildly dysmorphic facial features, which included small palpebral fissures (Figure 1c), simplified ears, a left accessory nipple, and a right club foot. Neurological examination revealed truncal and axial hypotonia, as well as brisk reflexes. Echocardiogram and ophthalmology evaluations were normal. The brain MRI, performed at the age of 19 months showed a thin corpus callosum (Figure 2c), incomplete myelination, and areas of signal abnormality in the central/periventricular white matter, partially involving the centrum semiovale (Figure 2d). These abnormalities were interpreted as secondary to gliosis. A focal asymmetric and larger area of signal abnormality, were associated with a small porencephalic cyst in the left caudate nucleus.
Patient 4
This male patient was the second child of non-consanguineous, healthy parents from South America. His sister is currently healthy. The patient was born at term of a pregnancy complicated by vaginal bleeding in the first trimester. His weight at birth was 3000 g (10–25th centile). Reportedly, the patient started walking at the age of 18 months. He lived in an orphanage between the ages of 18 months and 3 years, when he was adopted and moved to a different country. He was noted to exhibit communicative difficulties, hyperactivity, and lack of attention by the adoptive parents.
At the age of 3 years and 10 months his growth parameters were all within the normal range with a weight of 16 kg (50th centile), a height of 99 cm (25–50th centile), and a head circumference of 50 cm (25th centile). The patient had mild dysmorphic facial features (Figure 1d) and his physical exam was unremarkable. Additionally, he presented with severe attention deficits and borderline cognitive function with a score of 72 on the Griffiths scale. A brain MRI revealed a small arachnoid cyst. His EEG was normal and he had no history of seizures.
Patient 5
This male patient was the first child of healthy, non-consanguineous parents. He was born at term of an uncomplicated gestation weighing 4210 kg (>90th centile), with a length of 53.5 cm (>90th centile) and a head circumference of 36 cm (90th centile). At 5 months, he developed infantile spasms and developmental regression was noted. An EEG indicated the presence of hypsarrhythmia. Seizures improved during treatment with ACTH and topiramate. A follow-up EEG performed at 12 months showed improvement in the background rhythm, although there were bilateral anomalies in the temporal regions. Even after antiepileptics were discontinued, the patient did not experience another seizure.
On physical exam, he was normocephalic and non-dysmorphic (Figure 1e). On his last evaluation at the age of 3 years, his psychomotor development was delayed with severe speech involvement. He also exhibited perseverative, stereotyped hand movements as well as other behaviors characteristic of autistic spectrum disorders. A brain MRI uncovered an abnormal alteration of the white matter signal in the left temporal region.
Patient 6
This male, adult patient was evaluated for mental retardation and dysmorphic features. He was the second son of a non-consanguineous healthy couple. The patient's two brothers and one sister were, reportedly, in good health. The family history was negative for mental retardation, infantile spasms, and epilepsy. He was born at term of a gestation complicated by reduced fetal movements and the umbilical cord wrapping around his neck in utero. His birth weight was appropriate for the gestational age. He was noted to have a cleft palate and developed mild cyanosis at birth. From that point, his growth parameters remained around the third centile. His developmental delay was characterized by an inability to walk unsupported at the age of 4 years and a complete and ongoing lack of speech. The patient developed multiple tooth caries requiring removal of almost all of his teeth. At the age of 30 years, he was diagnosed with bilateral keratoconus. Before surgical correction of the keratoconus, he underwent general anesthesia, and subsequently went into cardiac arrest, which required cardiopulmonary resuscitation. A physical exam at the age of 33 years indicated his weight to be 52 kg (3rd–10th centile), his height 160 cm (below the 3rd centile), and his head circumference 53.5 cm (10–25th). His dysmorphic features included a high-frontal hairline, hyperplastic supra-orbital ridges, deep-set eyes, a prominent nasal bridge, mild asymmetric anteverted ears with mildly prominent anti-helix, a short philtrum, a thin upper lip, an everted lower lip, and micrognathia (Figure 1f). On eye examination, the bilateral keratoconus and a central leukoma involving the left eye were evident. A cardiac examination revealed a heart murmur, although an echocardiogram was not performed owing to a lack of patient compliance. He had cutaneous II–III syndactyly on the left foot. No focal signs were present on neurological exam and he was non-verbal and appeared to be severely mentally retarded. Because of his previous severe reaction to anesthesia, he was never evaluated with a brain MRI.
DECIPHER 248559
This patient was a 35-year-old male with severe mental retardation. He was the second of three children born to a couple from South America. By report, his younger brother had mild mental retardation. The patient's development was delayed and he developed behavioral problems after the first year of life. Poor eye contact, stereotyped behavior, severe anxiety, self-injuries, aggressive behavior, and sleeping problems were also particularly evident. At the age of 35 years, his speech was minimal and he was only able to say a few words.
His medical history was significant for constipation beginning in childhood, and for several hospital admissions related to severe constipation and pica. To that point, he had not developed seizures. At the age of 35 years, he weighed 73 kg at a height of 180 cm and a head circumference of 56 cm (−1SD). He was also observed to have a prominent jaw (Figure 1g), one irregular café-au-lait spot of ∼5 cm in diameter on the right side of his back, horizontal striae on his back, and short, large toes with a sandal gap. His fingers exhibited significant signs of self-inflicted lesions. A brain MRI was not performed.
Results
The results of the microarray CGH analyses in all seven cases examined are summarized in Table 1 and Figure 3. In patient 1, the CMA (BCM v7.2 OLIGO) showed a de novo 3.3–3.4 Mb microduplication on 14q12 between nucleotides 26 908 812–30 254 928. In patient 2, the array CGH analysis (BCM v7.0 OLIGO) revealed a 14q11.2q12 duplication of ∼9.4–11 Mb in size (minimal deleted interval 19 582 682–29 076 500), which was also detected by standard chromosome analysis. Chromosome analyses were normal in both parents. Metaphase and interphase FISH studies on 250 cells revealed that the duplicated chromosome 14 was present in 87% of the peripheral blood cultured lymphocytes analyzed. The mosaicism at a similar percentage of 86% was also detected by G-banded chromosome analysis on 105 metaphase cells.

Schematic representation of chromosome 14q11.2q13 duplications and the minimally duplicated region (gray shadow) in the seven presented cases (not drawn to scale). Each duplication is shown as a green bar. Benign CNV found in DECIPHER case 1366 is shown as a light blue bar. Triplication (from nucleotide 30 780–31 138 kb) found in patient 4 is shown as a pink bar and overlaps to a known benign copy number polymorphism (database of genomic variants: http://projects.tcag.ca/variation).16
In patient 3, the array CGH analysis (BCM V.6.5 OLIGO) revealed a gain in copy number in the 3q28q29 region spanning 7.6–8.4 Mb (minimal interval 191 656 626–199 287 624) as well as a gain in copy number in the 14q11.2q13.1 region, which spanned ∼13.7–18.4 Mb (minimal interval 19 582 682–33 275 612). Both chromosome and FISH analyses confirmed the presence of a supernumerary marker chromosome consisting of material from chromosomes 3 and 14. G-banded chromosome analysis of his parents showed the presence of a balanced translocation involving chromosomes 3 and 14 in his mother. Breakpoint sequencing of der(3)t(3;14)(q28;q13.1) in patient 3 located the breakpoints at 191 494 720(191 494 723) in chromosome 3 and 34 225 478(34 225 475) in chromosome 14. A 3 bp CTG microhomology was identified at the chromosome junction. The derivative chromosome 14 in the mother could not be studied because she declined to have her blood drawn.
In patient 4, conventional cytogenetic analysis also revealed the presence of a supernumerary marker chromosome present in 100% of the metaphases cells analyzed. Array CGH (180K, Agilent) revealed a duplicated and a triplicated region on 14q: first duplicated probe at 28 217 kb, first triplicated probe at 30 780 kb, last triplicated probe at 31 138 kb, last duplicated probe at 34 635 kb; flanking normal probes were at 28 191 kb and 34 648 kb. The triplicated region overlaps a copy number polymorphism detected in 2 out of every 50 individuals tested on a oligo CGH array16 and has been detected in array experiments using both the 44K and the 180K platforms, using different reference DNA. His mother's karyotype was normal, while the father could not be tested.
Array CGH analysis (180 K Agilent) on patient 5 revealed an ∼3 Mb duplication ranging from nucleotides 27 474 978–30 603 041. Normal parental CMA suggests that the duplication represents a de novo copy number change.
Analysis in patient 6 using a 44K Agilent array showed an ∼14.5 Mb duplication on 14q11.2–q13.1 between nucleotides 19 508 845–34 063 670. The same analysis in the parents of patient 6 revealed normal results.
A SNP array analysis in patient referred to as DECIPHER 248559 revealed a 14q11q12 duplication encompassing ∼6.79 Mb and extending between nucleotides 28 257 153 and 35 048 345.
The DECIPHER database includes another patient (DECIPHER 1366) with duplication in the 14q12 region, who presented with developmental delay, microcephaly, and dysmorphic features. Array CGH showed a 1.2 Mb gain in 14q12 that only includes the NOVA1 gene (RP11-330O19 to RP11-144C18; Figure 2). The patient's father, who carries the same duplication, was found to be phenotypically normal. Therefore, the copy number variant found in DECIPHER case 1366 is likely benign. Further investigation revealed that the patient is affected by Mowat–Wilson syndrome as confirmed by the presence of a frameshift mutation in the ZFHX1B gene.
Discussion
We report the clinical and molecular characterization of genomic duplications involving the 14q11.2q13.1 region in seven patients presenting with severe developmental and intellectual impairment, with major involvement of speech. Four of them (patients 1–3 and 5) also presented with developmental epilepsy, especially infantile spasms, as seen in patients 1, 3, and 5 (Table 2). Three of these patients (patients 1, 4, and 5) were non-dysmorphic, whereas the remaining four patients (patients 2, 3, 6, and 7) did not share any facial dysmorphisms (Figure 1).
The sizes of the interstitial 14q duplications detected in our series varied between ∼3–15 Mb. These duplications partially overlap the duplication found in the DECIPHER case 1366, which is thought to be a benign familial variant (Figure 2). The minimal duplicated region on 14q, common to patients 1–6 and to the DECIPHER 248559 case, includes only the following three genes: FOXG1, C14orf32, and PRKD1. The PRKD1 gene encodes a cytosolic serine–threonine kinase that binds to the trans-Golgi network and regulates the fission of transport carriers specifically destined for the cell surface.17 To date, no clinical phenotype has been associated with mutations or deletions of this gene. Although the FOXG1 gene represents the most interesting candidate to explain the abnormal neurodevelopmental phenotypes observed in our patients, the role of additional genes in the large sized duplicated regions cannot be excluded. For example, it has been proposed that the genes SUPT16H and CHD8 may account for the varying degrees of developmental delay/mental retardation in patients with 14q11.2 deletions18 and, thus, may also be relevant for patients with duplications involving this region. Nonetheless, these two genes are not duplicated in cases 1, 4, and 5, and DECIPHER 248559.
Moreover, we cannot rule out the phenotypic contribution of the 3q28q29 duplication in patient 3, whose supernumerary chromosome was likely the result of a 3:1 asymmetric segregation of the reciprocal balanced translocation present in his mother.19 The chromosomal abnormality (tertiary trisomy) resulted in duplications of 3q28qter and 14q11.2q13.1. However, 3q29 microduplications have been reported in asymptomatic parents of individuals with mild-to-moderate mental retardation and, therefore, its clinical significance is unclear.20, 21 Additionally, the seizures present in our case have not been reported in other patients with 3q29 microduplications.20, 21
FOXG1 has been shown to have an important role in the developing brain.22 It encodes a transcriptional repressor protein exclusively expressed in the fetal and adult brains.23 The mitotic expansion of Cajal–Retzius cells, the earliest neurons established in cortical layering, is suppressed by FOXG1.24 Supporting this notion, Foxg1 null mice have an excess of Cajal–Retzius neuron production in the cortex. Loss of function of the mouse gene FOXG1 also results in a severe reduction in the size of the cerebral hemisphere as well as early depletion of progenitor cells.25 FOXG1 also regulates neurogenesis in the postnatal hippocampus and continues to be expressed at high levels in areas of postnatal neurogenesis.26 Given its role in the developing brain, we hypothesize that increased FOXG1 dosage results in a clinical phenotype characterized by developmental delay and developmental epilepsy. Interestingly, overexpression of FoxG1 in chicken brain, via retroviral expression, resulted in thickening of the neuroepithelium and large outgrowth of the telencephalon and mesencephalum. This effect was not because of increased cell proliferation but rather a reduction in neuroepithelial apoptosis.27
Although MECP2 and 14q12 duplication can both lead to intellectual disabilities and absent or severely impaired speech,10 there are important differences. Developmental epilepsy – most specifically infantile spasms – was prominent in four of our seven cases, and in the single case previously reported,11 and to date, has not been described in MECP2 duplication patients. Interestingly, although epilepsy has been reported in patients with FOXG1 mutations and deletions, the onset of the seizures did not occur during infancy.1 Nevertheless, the similar phenotypes associated with MECP2 and FOXG1 losses and gains, the similar expression profiles of these genes in mouse postnatal forebrain tissue, as well as the overlapping subnuclear localization in neurons10 collectively support the hypothesis that these neurodevelopmental disorders are due to functional alterations in one or more common neuronal cell pathway(s).
In summary, we report a series of seven cases of patients with duplications in 14q involving the FOXG1 gene, who present with developmental delay/mental retardation and speech delay as predominant features, as well as developmental epilepsy in the majority of the patients, suggesting that increased dosage of FOXG1 is responsible for these abnormalities.
References
Ariani F, Hayek G, Rondinella D et al: FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83: 89–93.
Mencarelli M, Spanhol-Rosseto A, Artuso R et al: Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet 2009; 47: 49–53.
Papa FT, Mencarelli MA, Caselli R et al: A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am J Med Genet A 2008; 146A: 1994–1998.
Bisgaard AM, Kirchhoff M, Tumer Z et al: Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet A 2006; 140: 2180–2187.
Shoichet SA, Kunde SA, Viertel P et al: Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 2005; 117: 536–544.
Jacob FD, Ramaswamy V, Andersen J, Bolduc FV : Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet 2009; 17: 1577–1581.
Hagberg B, Hanefeld F, Percy A, Skjeldal O : An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett syndrome clinical criteria consensus panel satellite to european paediatric neurology society meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293–297.
Van Esch H, Bauters M, Ignatius J et al: Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–453.
Chahrour M, Zoghbi HY : The story of Rett syndrome: from clinic to neurobiology. Neuron 2007; 56: 422–437.
del Gaudio D, Fang P, Scaglia F et al: Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 2006; 8: 784–792.
Yeung A, Bruno D, Scheffer IE et al: 4.45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet 2009; 52: 440–442.
Firth HV, Richards SM, Bevan AP et al: DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009; 84: 524–533.
Cheung SW, Shaw CA, Yu W et al: Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 2005; 7: 422–432.
Ou Z, Kang SH, Shaw CA et al: Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med 2008; 10: 278–289.
Probst FJ, Roeder ER, Enciso VB et al: Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation. Am J Med Genet A 2007; 143A: 1358–1365.
de Smith AJ, Tsalenko A, Sampas N et al: Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet 2007; 16: 2783–2794.
Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K : PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem 1994; 269: 6140–6148.
Zahir F, Firth HV, Baross A et al: Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J Med Genet 2007; 44: 556–561.
Gardner RJM, Sutherland GR : Chromosome Abnormalities and Genetic Counseling. Oxford University Press: USA, 2004.
Ballif BC, Theisen A, Coppinger J et al: Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet 2008; 1: 8.
Lisi EC, Hamosh A, Doheny KF et al: 3q29 interstitial microduplication: a new syndrome in a three-generation family. Am J Med Genet A 2008; 146A: 601–609.
Hebert JM, McConnell SK : Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol 2000; 222: 296–306.
Murphy DB, Wiese S, Burfeind P et al: Human brain factor 1, a new member of the fork head gene family. Genomics 1994; 21: 551–557.
Hanashima C, Li SC, Shen L, Lai E, Fishell G : Foxg1 suppresses early cortical cell fate. Science 2004; 303: 56–59.
Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E : Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 1995; 14: 1141–1152.
Shen L, Nam HS, Song P, Moore H, Anderson SA : FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus 2006; 16: 875–890.
Ahlgren S, Vogt P, Bronner-Fraser M : Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol 2003; 57: 337–349.
Acknowledgements
PS was supported in part by Grant R13-0005-04/2008 from the Polish Ministry of Science and Higer Education. OZ is funded by the Cassa di Risparmio delle Provincie Lombarde (CARIPLO: 2007.5197, bando 2007) and by the Health Ministry (RF-AOM-2007-636538; ‘Genomic structural variation studies in mentally retarded and normal individuals in Italy’). MCB is funded by Telethon Grant GGP06208A. We thank Jessica Wu and Thao Quach for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Drs Brunetti-Pierri, Schaaf, Sutton, Xia, Cheung, and Stankiewicz are based in the Department of Molecular and Human Genetics at BCM, which offers genetic laboratory testing, including use of arrays for genomic copy number analysis, and derives revenue from this activity.
Rights and permissions
About this article
Cite this article
Brunetti-Pierri, N., Paciorkowski, A., Ciccone, R. et al. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet 19, 102–107 (2011). https://doi.org/10.1038/ejhg.2010.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.142
