
Abstract
So far, mutations in the human COL3A1 gene have been associated with the predominantly inherited Ehlers–Danlos syndrome (EDS), vascular type. Genotype–phenotype correlation perspectives collapsed, as haploinsufficiency, which was long suggested to confer a milder or unrecognized phenotype, was reported in four patients with a phenotype similar to that of vascular EDS. Here, we study a case of recessive EDS in a young consanguineous girl of healthy parents. She fulfilled the vascular EDS criteria for laboratory testing. Total sequencing of COL3A1 cDNA identified a homozygous nucleotide duplication (c.479dupT) resulting in a premature termination codon (p.Lys161GlnfsX45). Studies in genomic DNA showed that this mutation was inherited from each parent. The expression analysis (RT-PCR, quantitative-PCR, immunohistochemistry, WB) showed strong mRNA decay and an absence of type III collagen in the proband. The expected COL3A1 haploinsufficiency in her healthy ascendants did not lead to the manifestations of vascular EDS. This case provides evidence of a stochastic effect of COL3A1 haploinsufficiency in humans, which could be explained by the relation between nonsense-mediated mRNA decay efficiency and the resulting dominant-negative effect depending on the position of the mutation and/or modifying factors. It opens up new perspectives for the understanding of COL3A1 genotype–phenotype correlations, which is required while considering targeted therapy.
Similar content being viewed by others

Introduction
The Ehlers–Danlos syndrome (EDS) is defined by the triad: articular hypermobility, skin extensibility and tissue fragility. The vascular type (also known as type IV, MIM#130050, http://www.ncbi.nlm.nih.gov/Omim/)1 is the most severe type, with a median survival below 50 years of age in the largest cohort published to date.2 It is a rare predominantly inherited disorder, caused by mutations in the COL3A1 gene, which result in the synthesis of defective proα1(III) chains of type III procollagen.
The COL3A1 gene belongs to the highly homologous family of fibrillar collagens, which have several aspects in common:3 a triple-helical domain characterized by repeating Gly-X-Y triplets encoded by 43 exons (in COL3A1, exon 4 and 5 are fused in a single exon 4) that invariably begin with a glycine codon and have a similar pattern of size. Thus, the deletion of a single exon or splice-site mutation mostly results in an in-frame-shortened protein. A complex posttranslational processing with the removal of precursor-specific telopeptides and trimer units that further aggregate with other collagens results in ordered fibril structures and finally in periodic bundles of collagen fibers.4 Thus, because type III collagen is a homotrimer, the synthesis of an equal amount of chains from a normal and a mutated allele (if stable) predicts the assembling of a 7:1 ratio of abnormal/normal molecules through a dominant-negative effect, leading to a strong disorganization of collagen fibers. Almost all reported mutations in the COL3A1 gene (see the ‘database of human type I and III collagen mutations’ http://www.le.ac.uk/genetics/collagen/)5 match with this model. Although biases related to the screening methods for mutation detection are possible, this mutation spectrum, the analogy with the COL1A1 and COL1A2 mutations spectrum observed in osteogenesis imperfecta (MIM#120150 and 120160, http://www.ncbi.nlm.nih.gov/Omim/), and the findings from a mouse knockout model for COL3A16 have long suggested that COL3A1 null alleles could confer attenuated phenotypes. However, in 2001, the study by Schwarze et al7 questioned these genotype–phenotype correlations. In this study, four patients with a mutation described to result in haploinsufficiency had a clinical course similar to that in patients with classical missense or splice-site mutations. Consequently, the hope of an efficient gene-targeted therapy that could be based on the same premise as that of osteogenesis imperfecta8 collapsed. Here, we study a recessive case of vascular EDS observed in a consanguineous daughter of unaffected parents.
Participants and methods
Participants
The parents of the proband were uncle (II. 3) and niece (III.1). The pedigree is depicted in Figure 1. The proband (IV.3) was an 11-year-old Caucasian female. She was delivered by cesarean section at 36 weeks of gestation because of the premature rupture of membrane. Neonatal examination noticed normal parameters and bilateral clubfeet (talipes equinovarus). The ages at which she started to sit and walk were 15 and 30 months, respectively, and language acquisition was normal. She had Absence epilepsy, and her brain MRI revealed a diffuse cortical dysplasia with a predominantly frontal location and ventricular dilatation. A cardiac ultrasound showed a pulmonary valve stenosis. On account of easy bruising, a search for thrombopathy and coagulopathy was undertaken, which showed normal results. The proband came to our medical attention at the age of 10 years because of a clinical assessment suggesting EDS (Figure 1). She had thin, translucent skin with marked dystrophic scars, early-onset varicose veins and articular hypermobility, notably of small joints. Her growth parameters were 30 kg 700 (0SD); 133 cm (−0.5DS) and 54 cm (head circumference; +1DS). Her face was long with up-slanting palpebral fissures, slightly hypoplastic alae nasi and thin lips with a flat philtrum. She also exhibited multiple gingival recessions. A few weeks later, she presented acute abdominal pain related to small-bowel occlusion. Extreme intestinal, arterial and tissue fragilities were put to light during surgery that included the resection of approximatively 1 m of necrotic and perforated jejunum. Owing to hemorrhagic shock, an ileostomy by default was carried out. An attempt at restoration of bowel continuity was carried out after the first ileostomy failed. Three small-bowel perforations occurred during manual manipulation. She died 6 days after the intervention from laparotomy suture break down and total evisceration. The parents declined a post-mortem examination.

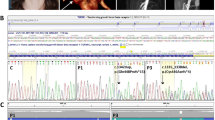
Pedigree, haplotype, clinical assessment and mutation status. (a) Haplotype study showing a large 56 cM chromosome 2 region (black boxed), inherited by descent in the proband (IV.3). Markers were purchased from Applied Biosystems, Courtaboeuf, France. Genotyping and analysis were carried out according to the manufacturer's instructions. Presence of the mutation, c.479dupT, is represented as a T in haplotype. (b) Characteristic Ehlers–Danlos syndrome (EDS) skin lesions of the Proband. (c) Electropherograms of COL3A1 exon 5 genomic sequences around the c.479dupT, and its detection with denaturing high-performance liquid chromatography (dHPLC, Transgenomic, Courtaboeuf, France) in genomic DNA (additional details in online-only material).
The familial investigation failed to record any suggestive signs or medical event that could suggest an EDS manifestation. The careful physical exam of both 43-year-old parents and the 13-year-old brother (IV.2) was totally negative for the Villefranche criteria.1 Premature death was traffic-related in participant II.1 and because of cancer in participants II.4 (throat) and III.2 (breast). The first born of the proband's parents (IV.1) died at 3 months of age from severe hypoxemia secondary to a diaphragmatic hernia (pathological data unavailable). No data were recorded for the common ancestral couple (participants I.1 and I.2).
Mutation analysis
Genetic and laboratory tests were carried out in the proband and her parents under conditions established by the French law, and appropriate written informed consents were collected. Blood samples were obtained from the proband and her parents. Skin fibroblasts and a frozen surgical sample of bowel (jejunum) were also collected from the proband. The parents refused skin biopsy. Human dermal fibroblasts (HDF) and a frozen jejunum sample were obtained as controls from the European Collection of Cell Culture (number: 06090715, 19 years old, Caucasian female) and from the Biological Resource Center of the Montpellier University Hospital (CHU Montpellier, France), respectively.
Total RNA was extracted using the RNeasy mini kit (Qiagen, Courtaboeuf, France) after fibroblasts were incubated for 24 h in the presence or absence of cycloheximide (CHX (100 μg/ml), Fluka, Buchs, Switzerland). Total RNA was converted into complementary DNA by means of an RT-PCR carried out by priming with random hexamers and SuperScript II Reverse transcriptase, according to the manufacturer's instructions (Invitrogen, Cergy-Pontoise, France). A control PCR was carried out to check for residual genomic DNA. Flexigene DNA and QIAamp DNA mini kits (Qiagen) were used to extract genomic DNA from blood and fibroblasts, respectively. Total cellular proteins were harvested from the cultured fibroblasts as described elsewhere.9
For mutation screening in the proband, all the coding sequences of the COL3A1 gene (cDNA) were sequenced bidirectionally in 10 overlapping fragments. For mutation confirmation in the proband and her parents, sequencing and denaturing high-performance liquid chromatography were carried out in genomic DNA. Chromosome 2 haplotype analysis was carried out to confirm biparental transmission. Finally, we carried out an expression analysis using RT-PCR, real-time quantitative PCR (LC-480, Roche, Manheim, Germany), immunohistochemistry and immunoblotting. Methods, conditions, primer sequences and antibody references are given in legends to figures and/or in online-only material. Exon numbering was carried out on the basis of the Reference Sequence NM_000090.3 (51 exons), as recommended by the Human Genome Variation Society guidelines.10 It is to be noted that, as stated above, our exon 4 may also be described as exon 4/5 in a historic numbering system (with 52 exons).
Results
COL3A1 transcript sequences of the proband were homozygous and showed a duplication in exon 5: c. 479dupT. This duplication led to a frameshift and a premature stop codon (PTC) in exon 7: p.Lys16fsGlnfsX45. It was found in the heterozygous state in the DNA of her healthy parents, validating the homozygosity found in the proband. Moreover, the proband was found homozygous by descent for a large 56cM portion of chromosome 2, between microsatellite markers, D2S306 and D2S126 (Figure 1). The predicted frameshift resulted in a sequence that did not contain a Gly-X-Y motif with a PTC at the very beginning of the triple-helical domain. To confirm that no collagen III could be synthesized in the proband, we performed several expression analysis experiments. First, the deposition of the RT-PCR products on 3% agarose gel showed that products were only detected in the proband after treatment with CHX (nonsense-mediated mRNA decay (NMD) inhibitor) (Figure 2). Second, real-time PCR and relative quantification analysis showed a normalized ratio of 1.133 (±0.211) in the HDF control in the absence of CHX, whereas this ratio was 0.0203 (±0.00155) in the proband (that is, COL3A1 transcripts were <2.5% of the control). In agreement with the first experiment, CHX exposure reestablished the expression level, with a normalized ratio of 1.728 (±1.034) and 0.733 (±0.18) in the control and proband, respectively (Figure 2). Third, western blotting showed no signal in the proband with an antibody against the N-terminal domain of the type III procollagen. Finally, the absence of collagen III synthesis in the proband was also sustained in situ by means of immunohistochemistry on jejunum samples (Figure 2).

Expression analysis in proband. (a) RT-PCR products separated on an agarose gel. Amplification of 25 cycles of COL3A1 exons 1–2 in the cDNA of a control individual (human dermal fibroblasts (HDF)) and the proband at different dilutions (1/10; 1/50 and 1/100) and with or without cycloheximide (CHX) treatment (only 1/50 dilution is shown). Although a specific transcript is detected in the control, none is detected in the proband without CHX treatment. (b) Real-time RT-PCR. PCR products of COL3A1 exons 5–8 amplicon (primers in online only material) were quantified using the LightCycler 480 system with SYBR Green I Master Mix according to the manufacturer's instructions (Roche, Mannheim, Germany). The expression level of COL3A1 was normalized to that of housekeeping genes, β-actin and GAPDH (Eurogentec, Angers, France). A calibrator sample (HDF) was used and experiments were repeated thrice. In each experiment, the samples were run in triplicates. The COL3A1 expression ratio was calculated using the E-Method (Roche, Eurogentec, Angers, France). (c) Western blot analysis: Immunodetection of Type III procollagen by an antibody against the N-telopeptide (S17, Santa Cruz, Santa Cruz, CA, USA) and by an antibody against the reference gene, β-actin (AC-15, Santa Cruz). We loaded 25, 40 and 60 μg of total proteins that were extracted from both the medium and the cell layer of the skin-cultured fibroblast from the proband and from the control. No (pro)collagen III was detected in the proband. (d) Immunohistochemistry: in situ detection of (pro)collagen III in frozen jejunum samples of the proband and control (48 years old, Caucasian female). In the proband, only autofluorescence of the connective tissue is detected after labeling with the S17 antibody (Santa Cruz), whereas the control showed intense labeling (concordant data were obtained with antibodies against the full-length collagen III triple-helical domain (FH-7A, Santa Cruz; data not shown)).
Discussion
Mutations that confer an unusual pattern of inheritance in a gene related to a well-known genetic disease can sometimes highlight a particular mechanism that is useful for correlating genotype to phenotype. For example, the efficiency of the NMD pathway can alter the pattern of inheritance in several genetic disorders.11 We report here the characterization of a recessive inheritance of EDS linked to a COL3A1 null allele, inherited by descent in a consanguineous child. Recessive inheritance of EDS type IV has been suggested in the past.12 However, there were not enough data to discriminate real recessive cases from de novo occurrences, mosaicisms or even from marked phenotypic variability. It is interesting that the proband did not present a typical vascular-type EDS phenotype. First, she had diffuse cortical dysplasia and absence of epilepsy. This could suggest a phenotypic overlap with the EDS variant caused by mutations in the gene encoding Filamin A (MIM#300537). Moreover, this variant, as well as the vascular type, includes cardiovascular manifestations. However, the proband's MRI of the brain showed no periventricular nodular heteropia (a hallmark of this EDS variant). Considering the fact that numerous etiologies remain possible (including recessive disease), this overlap has to be interpreted with caution. Second, she had pronounced atrophic scars such as those found in classical EDS type. However, there are striking parallels between our observation and that in the mouse knockout model described by Liu et al.6 Homozygous mutant mice had a low-survival rate, with most deaths occurring within 2 days after birth. This could be consistent with the premature death of the proband's sister. The surviving homozygous mutant mice lived one-fifth of the normal life span. In addition to aneurysms, they displayed marked skin lesions, frequent intestinal enlargement and occasionally fatal intestinal rupture. This correlates with our observation in humans (although we cannot exclude another gene defect), suggesting a distinct phenotype for recessive inheritance. Finally, similar to heterozygous mice, the heterozygous parents were phenotypically normal. We could not extend the familial investigation to other relatives, however no evident vascular EDS manifestations were recorded. Therefore, it is likely that the common ancestor and potentially other relatives carrying the mutation also match the mouse model. Thus, heterozygous as well as homozygous individuals for COL3A1 null alleles could present a distinct and unrecognized phenotype. Taken together, these points also support the classic dominant-negative mechanism as being the main basis for genotype–phenotype correlations in vascular EDS.
How could we conciliate these findings with the study of Schwarze et al?7 It is unlikely, even if possible, that the authors failed to detect a mutation in trans in their reported patients (non-detected recessive cases). Therefore, two hypotheses remain to explain the discrepancy.
The first hypothesis refers to the relation between NMD efficiency and the resulting dominant-negative effect. Our findings, as well as those of Schwarze et al,7 present evidence of a strong mRNA decay by NMD. If admitted that NMD is rarely, if ever, 100% efficient,13 the pathogenicity of the mutation would be linked to its position. In their study, c.1832_1833delAA and c.555delT mutations occurred in the Gly-X-Y triple-helical domain. If translated, the little remnant peptide could be sufficient to alter the complex post-transductional processes of the type III collagen through a dominant-negative effect. It is also consistent with their findings regarding the c.4294C>T mutation, which result in a truncated peptide (PTC in the last COL3A1 exon). Finally, another explanation is necessary to modelize the effect of the remaining c.413delC mutation. Indeed, this mutation, similar to the c.479dupT mutation reported here, also predicts an absence of the Gly-X-Y major triple-helical domain. However, c.413delC also disrupts the Gly-X-Y repeats of the minor triple-helical domain of the N-telopeptide, which is known to be required for N-terminal proteinase recognition and cleaving.14 This is not the case with c.479dupT, which occurs in the microunfolding region (last part of the N-terminal domain). c.413delC could thus lead to a stronger interference with N-propeptide removal and this may explain the phenotype discordances.
The second hypothesis would be that (a) modifier gene(s)/factor(s) confer(s) a protective effect in the family described here. Such a mechanism has been documented to explain the marked variability of symptoms in other diseases. For example, for a same mutation in the COL7A1 gene, different alleles of a frequent functional variant in the gene encoding the matrix-metalloproteinase 1 (also known as collagenase) have been described to produce different forms of recessive dystrophic epidermolysis bullosa.15 Such modifier genes could also support the marked variability observed in some vascular EDS pedigrees and in the discrepancy between haploinsufficiency consequences in the Schwarze study and in ours. Cis-acting elements could be more easily identified if there is a correlation between phenotype and variations in the expression of the wild-type allele. However, up to now, we have had no fibroblast cultures of the clinically unaffected heterozygous parents. It is interesting that these two hypotheses are not exclusive. Recently, NMD efficiency has been shown to vary between cell types, tissues and individuals with the growing idea that the NMD pathway could act as a modulator of genetic disease severity.16
The mechanisms by which mutations in the COL3A1 gene produce diseases are still poorly understood. So far, all the mutations reported in COL3A1 have been linked to an autosomal dominant pathway of inheritance. For the first time, we have described herein a recessive EDS case caused by an autozygous nonsense mutation in the COL3A1 gene. This observation opens up new perspectives for genotype–phenotype correlations. Understanding why heterozygosity for a COL3A1 null allele can result in an apparently normal phenotype in a mouse and, here, in humans could offer seductive therapy perspectives for this life-threatening disease.
References
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ : Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Am J Med Genet 1998; 77: 31–37.
Pepin M, Schwarze U, Superti-Furga A, Byers PH : Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Prockop DJ, Kivirikko KI : Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 1995; 64: 403–434.
Myllyharju J, Kivirikko KI : Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 2004; 20: 33–43.
Dalgleish R : The human collagen mutation database 1998. Nucleic Acids Res 1998; 26: 253–255.
Liu X, Wu H, Byrne M, Krane S, Jaenish R : Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA 1997; 94: 1852–1856.
Schwarze U, Schievink WI, Petty E et al: Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet 2001; 69: 989–1001.
Prockop DJ : Targeting gene therapy for osteogenesis imperfecta. N Engl J Med 2004; 350: 2302–2304.
Rene C, Taulan M, Iral F et al: Binding of serum response factor to cystic fibrosis transmembrane conductance regulator Carg-like elements, as a new potential CFTR transcriptional regulation pathway. Nucleic Acids Res 2005; 33: 5271–5290.
den Dunnen JT, Antonarakis SE : Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15: 7–12.
Khajavi M, Inoue K, Lupski JR : Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 2006; 14: 1074–1081.
Pope FM, Martin GR, McKusick VA : Inheritance of Ehlers-Danlos type IV syndrome. J Med Genet 1977; 14: 200–204.
Isken O, Maquat LE : Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev 2007; 21: 1833–1856.
Cabral WA, Makareeva E, Colige A et al: Mutations near amino end of alpha 1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem 2005; 280: 19259–19269.
Titeux M, Pendaries V, Tonasso L, Decha A, Bodemer C, Hovnanian A : A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum Mutat 2008; 29: 267–276.
Viegas MH, Gehring NH, Breit S, Hentze MW, Kulozik AE : The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the Nonsense Mediated Decay pathway. Nucleic Acids Res 2007; 35: 4542–4551.
Acknowledgements
We gratefully acknowledge S Auvin, B Catteau, S Coopman, JM Cuisset and R Sfeir for referring the patient and for providing follow-up information, C Baudoin, N Pallares-Ruiz, C Rene and M Taulan for their technical advice and support and N Hornez for helping in preparing the paper. We also thank the clinical research team of the CHU of Montpellier, notably J Dinet and S Plagnol, for their support. A Plancke and part of this work were funded by the ‘Programme de Soutien aux Techniques Innovantes Coûteuses 2006’ from the French Ministry of Health. We are also especially grateful to X Jeunemaitre for his helpful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Plancke, A., Holder-Espinasse, M., Rigau, V. et al. Homozygosity for a null allele of COL3A1 results in recessive Ehlers–Danlos syndrome. Eur J Hum Genet 17, 1411–1416 (2009). https://doi.org/10.1038/ejhg.2009.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.76
Keywords
This article is cited by
-
Variant-specific effects define the phenotypic spectrum of HNRNPH2-associated neurodevelopmental disorders in males
Human Genetics (2022)
-
The Ehlers–Danlos syndromes
Nature Reviews Disease Primers (2020)
-
Understanding the basis of Ehlers–Danlos syndrome in the era of the next-generation sequencing
Archives of Dermatological Research (2019)
-
Vascular Ehlers–Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family
European Journal of Human Genetics (2015)
-
GPR56 and the Developing Cerebral Cortex: Cells, Matrix, and Neuronal Migration
Molecular Neurobiology (2013)
