
Abstract
The lipoprotein lipase (LPL) gene encodes a rate-limiting enzyme protein that has a key role in the hydrolysis of triglycerides. Hypertriglyceridemia, one widely prevalent syndrome of LPL deficiency and dysfunction, may be a risk factor in the development of dyslipidemia, type II diabetes (T2D), essential hypertension (EH), coronary heart disease (CHD) and Alzheimer's disease (AD). Findings from earlier studies indicate that LPL may have a role in the pathology of these diseases and therefore is a common or shared biological basis for these common complex diseases. To examine this hypothesis, we reviewed articles on the molecular structure, expression and function of the LPL gene, and its potential role in the etiology of diseases. Evidence from these studies indicate that LPL dysfunction is involved in dyslipidemia, T2D, EH, CHD and AD; and support the hypothesis that there is a common or shared biological basis for these common complex diseases.
Similar content being viewed by others
Introduction
The lipoprotein lipase (LPL) gene is located on 8p22, spans ∼30 kb and contains 10 exons and encodes a rate-limiting enzyme called lipoprotein lipase. LPL has a key role in the hydrolysis of triglycerides (TG). In 1960, LPL deficiency was discovered by Havel and Gordon,1 and several mutations were identified upon cloning the gene in 1991 by Henderson et al.2 Subsequently, more subtle mutations were detected in the LPL gene, and some mutants are reputed to influence LPL activity and has a role in the disease's etiology.2, 3 LPL is one member of the triglyceride (TG) lipase gene family, which includes pancreatic lipase (PL), hepatic lipase (HL) and newly discovered endothelium lipase (EL).4 It is a rate-limiting enzyme in the hydrolysis of triglyceride-rich particles,5 such as chylomicron and very-low-density lipid (VLDL), thereby providing non-ester acids and 2-monoacylglycerol for tissue use.6 Recently, in addition to hydrolysis, LPL has been reported to be involved in lipid intake and clearance.7, 8, 9, 10 Variations in LPL gene sequence, expression and regulation may influence its function and contribute to diseases. The changes in LPL activity in adipose and muscle tissues suggest that LPL is regulated in a tissue-specific manner.4, 11, 12 Several allelic variants were reported to affect the expression and activity of LPL. In this paper, we review the literature relating to LPL focusing on five diseases, namely dyslipidemia, type II diabetes (T2D), essential hypertension (EH), coronary heart disease (CHD) and Alzheimer's disease (AD) to see whether there is any common etiological mechanism involving LPL.
LPL and dyslipidemia
LPL is the key enzyme in the hydrolysis of triglycerides packaged in chylomicron and VLDL, thereby providing non-festered fatty acids (NEFA) and 2-monoacylglycerols for many tissues.8, 9 In adipose, NEFA is re-esterified for storage in the form of triacylglycerol (TAG); but in muscles, oxidation of NEFA is the major source of energy.6 According to these studies above, it might be reasonable to considering LPL as the preferred candidate gene for the dyslipidemia.
The relationship between LPL and dyslipidemia is very well established and independent of ethnic background.1, 2, 3, 13 Several clinical observations have suggested that the patients with LPL deficiency suffer severe hypertriglyceridemia.3, 14 The genetic variants of LPL may have a role in determining lipid levels. A study by Jemaa et al.15 in French patients (614 patients with myocardial infarction and 733 controls; P<0.01) indicated that there is an association between LPL rs328 C → G (Ser447Stop) polymorphism and hypertriglyceridemia. Their result is supported by Groenemeijer et al.16 study in Dutch patients (820 patients of coronary artery disease; P=0.044), and by King et al. (1998) finding17 and Yamada et al.18 report (in 5213 Japanese individuals; P=0.0007). The above results are also supported by evidence from the Chinese samples. A genetic screen of LPL gene of 53 hypertriglyceridemia patients in 26 T2D Chinese pedigrees identified that three novel mutations, Lys312insC, Thr361LnsA and double mutations Lys312insC+Asn291Ser, were clinically associated with hypertriglyceridemia, which was further confirmed by the mutagenesis with significantly reduced LPL activity (P<0.01) and expression studies.19 Yang et al.20 found that two SNPs HindIII (rs320) and HinfI (rs328) at LPL were associated with cholesterol levels (P=0.0178 and P=0.0088, respectively) in Chinese cohort.
This association is further validated by LPL target therapy studies. Ross et al.21 observed that intramuscular administration of an adeno-associated virus serotype 1 (AAV1) vector encoding the human LPLS447X variant cDNA (AAV1-LPLS447X) could normalize the dyslipidemia in LPL−/− mice, and Kodera et al. reported that anti-LPL autoantibody could elevate serum triglyceride levels by inhibiting 47% activity of LPL (P<0.0001).22
LPL and T2D
T2D is widely prevalent in the world's populations and it is predicted that the prevalence will increase to 5.4% by the year 2025.17 Both hereditary and environmental factors contribute to individual susceptibility; however, dysfunction of LPL may explain, at least in part, the prevalence of T2D.
Hölzl et al.23 reported a study that enrolled 85 heterozygous carriers of a missed (Gly188Glu) mutation or a splice site mutation (c-in position-3 at the acceptor splice site of intron 6) in the LPL gene and 108 non-carriers. Their results indicated that the heterozygous carriers had elevated triglyceride level and reduced insulin sensitivity compared with non-carriers (P<0.0005 and P=0.014, respectively). A meta-analysis including 19 246 individuals showed nominal significant association between LPL N291S (rs268) variant and T2D (odds ratio=2.26, 95% confidence interval (CI): 1.02–4.99, P=0.04) and CHD (odds ratio=1.48, 95% CI: 1.09–2.00, P=0.01). Although not convincing, together with the powerful correlation between significantly decreased catalytic activity (∼60% of the wild type) and secretion ability (about 50% of the wild type) of N291S mutant and deleterious lipid profile (triglycerides: 32.3% increase and HDL-C: 34.2% decrease), which are risk factors of T2D and CHD, this association may be not a false-positive result and further study is necessary and justifiable.24 Goodarzi et al.25 analyzed haplotypes of six polymorphisms in LPL and provided compelling evidence that the LPL gene might have a role in determining insulin sensitivity (P=0.031, haplotype 1: GATTCG) and insulin resistance (P=0.007, haplotype 4: GAGGGG) in their Mexican-American population. Transgenic (Tg) watanabe heritable hyperlipidemic (WHHL) rabbits that overexpress the human LPL gene were generated by Koike et al. In addition to dramatically ameliorating hypertriglyceridemia and hypercholesterolemia, overexpression of LPL was able to suppress high-diet-induced obesity and insulin resistance in these Tg WHHL rabbits.26 However, two reports27, 28 suggest that overexpression of human LPL in skeletal muscle of mouse could aggravate insulin resistance through the insulin signal cascade. A recent study found that the reduced LPL delivery in skeletal muscle in skeletal muscle-specific LPL knockout mouse (SMLPL−/−) induced increased insulin sensitivity in skeletal muscle but insulin resistance in other metabolic tissues, and ultimately led to obesity and systematic insulin resistance.29 These results suggested that systematic overexpression of LPL might improve insulin resistance, although the underlying mechanism is not clear. These controversies suggest that the association between the LPL gene and T2D may be tissue specific. Some results relevant to the underlying mechanisms have been reported as follows: Cruz et al.29 study indicated that more FFA could be delivered to pancreatic β-cells by increasing LPL activity, consequently impairing β-cell function and promoting apoptosis in the patients with hyperglyceridemia, hyperinsulinemia and T2D. Hypertriglyceridemia prioritizing the utilization of TAG as fuels inhibits the intake and oxidization of glucose;27 intracellular fatty acid metabolites interfere with propagation of insulin signaling cascade;28 and delivering more FFA to pancreatic β-cells impairs β-cell function and promote apoptosis.29 The above findings may be partly responsible for the association between LPL and T2D.
LPL and EH
The occurrence of EH is affected by hereditary and environmental factors and interaction between them.30 Many previous findings have shown that abnormal lipid metabolism and insulin resistance may have a role in the process of EH. LPL, hydrolyzing TG and regulating lipid metabolism, is logical to be regarded as the candidate gene for EH.
Linkage analysis of 148 Chinese hypertensive pedigrees with seven micro-satellite makers of LPL gene discovered links between systolic blood pressure (SBP) and marker D8S261 (LOD=2.68) and NEFL (LOD=2.1), between diastolic blood pressure (DBP) and marker D8S1145 (LOD=1.06) and NEFL (LOD=1.22) in the region of 8p22 (LPL located on the region); two common mutations HindIII (rs320; P=0.004) and HinfI (rs328; P=0.004) in the LPL gene were both associated with DBP.31 In addition, another quantitative-transmission/disequilibrium test (TDT) also showed significant association between EH and D8S261 (P=0,0002 and 0.033 for SBP and DBP, respectively).32 The association of LPL and hypertension was supported by another independent study in the Chinese Han population, indicating that a polymorphism in intron 8 of PLP may be a risk variant for EH (OR=2.9, P<0.01).33 These findings have not been replicated in Caucasian samples34 and suggest that LPL might have a race-specific role in the development of hypertension. The underlying mechanisms might include the effect of elevated TG on hemodynamics, sodium re-absorption, retention and vascular hypertrophy,15 and the role of LPL in the regulation of arterial stiffness.31
LPL and CHD
It is known that insulin resistance and hypertension are independent risk factors for CHD. On the basis of the linkage of the LPL gene to these factors, LPL is a candidate gene for CHD. A study by Socquard et al.35 in 404 unrelated French patients with T2D found that the prevalence of CHD in this population was 18%, but increased to 29 and 38% in H2H2 (rs320, the site of HindIII) and P2P2 (rs285, the site of PvuII), respectively, polymorphism carriers at LPL (P< 0.02). The findings by Zee et al.36 indicated that LPL N291S (rs268) genetic variant is an independent predictor of increased risk of venous thromboembolism (OR=3.09; 95% CI: 1.56–6.09; P=0.001), also supporting a role for the LPL gene in vascular biology. One study among 2484 inhabitants of the municipality of Hoorn discovered that low LPL activity and high triglyceride concentration are determinants of small LDL size,37 which is associated with increased risk of cardiovascular disease (CVD).38 The naturally occurring LPL S447X, which was shown to increase the lipolytic function of LPL and a gain-of-function mutation, could reduce the CVD risk compared with S447S.39 All of these observations suggest that LPL might involve in the etiology of CHD. However, the underlying mechanism is complicated and tissue specific. Overexpression of LPL in monocyte-derived macrophages (MDM) induces unregulated uptake of NEFA and 2-monoacyglycerol and re-esterified into TAG. The lipid saturated macrophages evolve into foam cells and then penetrate the endothelium cells into the middle layer of vascular wall.40 In addition, the LPL could trigger other pro-pathogenic events, such as the proliferation of smooth muscle cells.41 A marked decreases in diet-induced atherosclerosis was detected in chimerical mice that are deficient for macrophage LPL expression,42 and atherosclerosis was accelerated in transgenic apoE-deficient mice by macrophage-specific expression of human LPL.43 In the adipose and muscle tissues, however, LPL acts protectively because it aids in the clearance of circulating lipoprotein particles through storage or utilization.6
LPL and AD
Recently, reports on the association between LPL and AD have increased. No significant distribution difference (P=0.09) of S477X (rs328) is found between 852 patients with AD and 190 healthy controls in Baum et al. study,44 which is supported (P>0.05) by the Fidani et al. report.45 However, Blain et al.46 study (in 153 patients with AD and 242 controls from Canada), on a PvuII SNP polymorphism (rs285) of LPL, discovered that homozygote of P+ allele resulted in an odds ratio of 2.3 for the risk of developing AD; the P+ allele could significantly affect its mRNA expression level (P=0.026), brain tissue cholesterol levels (P=0.0013), neuro-fibrillary tangles (P=0.025) and senile plaque (P=0.022) densities. In 243 Italian patients with AD and 148 healthy subjects, the H+ allele of HindIII SNP (rs320) showed a trend but not strong association with AD, the odds ratio was 2.7 (95% CI: 1.01−7.21; P=0.048); in addition, the homozygote of H+H+ genotype could increase the risk of developing AD (P=0.029).47 These discrepancies may be attributed to one of the following reasons. First, it is possible that different ethnic groups may have different risk alleles in the same gene or may have different predisposing genes for AD. Second, it is possible that an independent study with small samples has limited power to detect association of small effect, and is prone to false positives or negatives. Finally, false positives or negatives owing to statistical chance cannot be completely eliminated.
Findings from the neuroblastoma cells suggest that LPL expression in neurons stimulates the extension of neuritis and protects the neurons against oxidized lipoproteins toxicity.48 LPL overexpression was found in the lesion brain region in mouse models, and LPL mRNA level was upregulated in microglial cells during ischemia produced by occlusion of the middle cerebral artery, and was involved in the scavenging and recycling of lipids by microglial cells in the brain.49, 50 Thus, LPL secreted by microglial cells may not only help in the recycling of lipids but could also potentially act as a trophic factor for neuronal survival and differentiation. AD is considered as a neurodegenerative disease, however, and multiple pieces of evidence show that oxidative stress may also have a pathogenic role in AD.51 These results suggest that LPL might have a role in the antioxidation stress of brain through lipids scavenging and recycling, bonding function and other unrecognized mechanisms, and indicate that LPL may be a protective factor of AD. Therefore, dysfunction of antioxidation stress induced by LPL aberration, in the brain, might be a risk factor of AD. More investigations of genetics and biology are required to confirm the association between LPL and AD, and to uncover the mechanism of LPL underlying AD.
Discussion
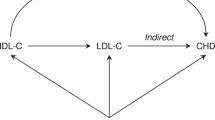
We have shown the existence of a common biological basis for several common complex diseases. The examples given here support the idea that LPL is involved in several diseases such as dyslipidemia, T2D, EH, CHD and AD. Hypertriglyceridemia, T2D, EH, CHD and AD, all display dyslipidemia in the initial stage or throughout their whole pathological process. Owing to the lipolytic function of LPL, it was considered a good candidate gene for these diseases. Catalytic function of LPL is the main basis in these relationships, deficient synthesis or dysfunction of LPL reduces hydrolysis of chylomicron and VLDL, hindering intake of lipids, and eventually resulting in redundant accumulation of lipoproteins in the plasma.6 The antioxidation stress function of LPL promotes clearance of the lipid remnants from the circulation, which are re-esterified for storage and utilization in tissues.7, 8, 9, 10, 48, 49, 50 Dyslipidemia, induced by the aberration of the LPL gene, might be the shared intermediate process on the development of these five diseases. The hypertriglyceridemia could directly be induced by the dysfunction of LPL. However, T2D, EH, CHD and AD are indirectly associated with LPL by different patho-physiological mechanisms and signal pathways (see Figure 1). According to the findings reported here, we surmise that LPL's antioxidation stress may be one important mechanism underlying the etiology of T2D, EH, CHD and AD, but more research is required to elucidate this. Therefore, dyslipidemia and dysfunction of antioxidation stress induced by aberration of LPL gene might be two risk factors for the development of T2D, EH, CHD and AD.

The schematic of the relationships among the lipoprotein lipase (LPL) gene and dyslipidemia, type II diabetes (T2D), essential hypertension (EH), coronary heart disease (CHD) and Alzheimer disease (AD). Dyslipidemia, induced by the aberration of the LPL gene, might be the shared intermediate process on the development of these five diseases. Hypertriglyceridemia features dyslipidemia. Involves in regulating the pancreatic β-cell function, β-cell apoptosis and the insulin signal cascades, which aggravates insulin insufficient secretion and insulin resistance and then induces T2D. Hyperinsulinemia induced by elevated TG, may be partly responsible for EH by its functions on renal hemodynamics, sodium re-absorption, retention and vascular hypertrophy. Elevated TG is associated with atherosclerosis and thromboembolism; both of them are high-risk factors for the development of CHD. LPL has a role in the antioxidation stress of brain through lipids scavenging and recycling, regarded as a protective factor for Alzheimer's disease (AD). Dysfunction of antioxidation stress induced by LPL aberration, in the brain, might be a risk factor of AD.
Although evidence from the LPL gene studies supports the hypothesis that there is a common biological basis for the common complex diseases, a number of questions have also arisen from these studies that need to be investigated in more detail. First, the molecular mechanisms that potentially link LPL to T2D, EH, CHD and AD need specific attention in future research. Second, even for a widely examined polymorphism, such as S447X (rs328), reports from different ethnic samples are not consistent and studies based on large and well-matched samples are required. Third, as described in this review, tissue/cell-specific regulation of LPL has a major implication for diseases, such as T2D and CHD; and this selective modulation of LPL function is important for understanding its pathological mechanism and finding new therapeutic techniques. Fourth, an interesting issue for the future studies is whether or not LPL might have a part in the development of other diseases such as schizophrenia.52
In short, evidence from these studies of LPL are consistent with that from Rzhetsky et al.53 support the idea that separate genetic diseases can overlap in their pathogenesis, especially for diseases that share similar clinical phenotypes. As phenotypic overlap is a good predictor of functional relatedness of the underlying susceptibility genes or molecular mechanisms,54 these results indicate that one gene associated with multiple diseases might exist in the population and this phenomenon should be considered as a network of interrelated diseases and disease traits, rather than a list of distinct disease entities.55 The detailed and accurate exploration of this interesting and important hypothesis should be helpful for elucidating the etiology of common complex diseases, and has a potential impact on the study for liability genes of complex human diseases.
References
Havel RJ, Gordon Jr RS : Idiopathic hyperlipemia: metabolic studies in an affected family. J Clin Invest 1960; 39: 1777–1790.
Henderson HE, Ma Y, Hassan MF et al: Amino acid substitution (Ile194—Thr) in exon 5 of the lipoprotein lipase gene causes lipoprotein lipase deficiency in three unrelated probands. Support for a multicentric origin. J Clin Invest 1991; 87: 2005–2011.
Liu A, Lee L, Zhan S et al: The S447X polymorphism of the lipoprotein lipase gene is associated with lipoprotein lipid and blood pressure levels in Chinese patients with essential hypertension. J Hypertens 2004; 22: 1503–1509.
Creyer A : Tissue lipoprotein lipase activity its action in the lipoprotein metabolism. Int J Biochem 1981; 13: 525–541.
Wang CS, Hartsuck J, McConathy WJ : Structure and functional properties of lipoprotein lipase. Biochim Biophys Acta 1992; 1123: 1–17.
Seip RL, Semenkovich CF : Skeletal muscle lipoprotein lipase: molecular regulation and physiological effects in relation to exercise. Exerc Sport Sci Rev 1998; 26: 191–218.
Merkel M, Kako Y, Radner H et al: Catalytically inactive lipoprotein lipase expression in muscle of transgenic mice increase very low density lipoprotein uptake: direct evidence that lipoprotein lipase bridging occurs in vivo. Proc Natl Acad Sci USA 1998; 95: 13841–13846.
Merkel M, Heeren J, Dudeck W et al: Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J Biol Chem 2002; 277: 7405–7411.
Mead JR, Ramji DP : The pivotal role of lipoprotein lipase in atherosclerosis. Cardiovasc Res 2002; 55: 261–269.
Mamputu JC, Desfaits AC, Renier G : Lipoprotein lipase enhances human monocyte adhesion to aortic endothelial cells. J Lipid Res 1997; 38: 1722–1729.
Doolittle MH, Ben-Zeev O, Elovson J et al: The response of lipoprotein lipase to feeding and fasting. Evidence for posttranslational regulation. J Biol Chem 1990; 265: 4570–4577.
Lithell H, Boberg J, Hellsing K et al: Lipoprotein lipase activity in human skeletal muscle and adipose tissue in the fasting and the fed states. Atherosclerosis 1978; 30: 89–94.
Wittrup HH, Tybjærg-Hansen A, Nordestgaard BG : Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease: a meta-analysis. Circulation 1999; 99: 2901–2907.
Feoli-Fonseca JC, Levy E, Godard M et al: Familial lipoprotein lipase deficiency in infancy: clinical, biochemical, and molecular study. J Pediatr 1998; 133: 417–423.
Jemaa R, Fumeron F, Poirier O et al: Lipoprotein lipase gene polymorphisms: associations with myocardial infarction and lipoprotein levels, the ECTIM study. J Lipid Res 1995; 36: 2141–2146.
Groenemeijer BE, Hallman MD, Reymer PW et al: Genetic variant showing a positive interaction with β-blocking agents with a beneficial influence on lipoprotein lipase activity, HDL cholesterol, and triglyceride levels in coronary artery disease patients: the Ser447-stop substitution in the lipoprotein lipase gene. Circulation 1997; 95: 2628–2635.
King H, Aubert RE, Herman WH : Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 1998; 21: 1414–1431.
Yamada Y, Matsuo H, Warita S et al: Prediction of genetic risk for dyslipidemia. Genomics 2007; 90: 551–558.
Hu Y, Ren Y, Luo RZ et al: Novel mutation of the lipoprotein lipase gene associated with hypertriglyceridemia in members of type 2 diabetes pedigrees. J Lipid Res 2007; 48: 1681–1688.
Yang W, Huang J, Yao C et al: Linkage and linkage disequilibrium analysis of the lipoprotein lipase gene with lipid profiles in Chinese hypertensive families. Clin Sci (Lond) 2005; 108: 137–142.
Ross CJ, Twisk J, Meulenberg JM et al: Long-term correction of murine lipoprotein lipase deficiency with AAV1-mediated gene transfer of the naturally occurring LPL (S447X) beneficial mutation. Hum Gene Ther 2004; 15: 909–919.
Kodera M, Hayakawa I, Komura K et al: Anti-lipoprotein lipase antibody in systemic sclerosis: association with elevated serum triglyceride concentrations. J Rheumatol 2005; 32: 629–636.
Hölzl B, Iglseder B, Sandhofer A et al: Insulin sensitivity is impaired in heterozygous carriers of lipoprotein lipase deficiency. Diabetologia 2002; 45: 378–384.
Hu Y, Liu W, Huang R, Zhang X : A systematic review and meta-analysis of the relationship between lipoprotein lipase Asn291Ser variant and diseases. J Lipid Res 2006; 47: 1908–1914.
Goodarzi MO, Guo X, Taylor KD et al: Lipoprotein lipase is a gene for insulin resistance in Mexican Americans. Diabetes 2004; 53: 214–220.
Koike T, Liang J, Wang X et al: Overexpression of lipoprotein lipase in transgenic watanabe heritable hyperlipidemic rabbits improves hyperlipidemia and obesity. J Biol Chem 2004; 279: 7521–7529.
Ferreira LD, Pulawa LK, Jensen DR et al: Overexpressing human lipoprotein lipase in mouse skeletal muscle is associated with insulin resistance. Diabetes 2001; 50: 1064–1068.
Pulawa LK, Eckel RH : Overexpression of muscle lipoprotein lipase and insulin sensitivity. Curr Opin Clin Nutr Metab Care 2002; 5: 569–574.
Cruz WS, Kwon G, Marshall CA et al: Glucose and insulin stimulate heparin-releasable lipoprotein lipase activity in mouse islets and INS-1 cells. A potential link between insulin resistance and beta-cell dysfunction. J Biol Chem 2001; 276: 12162–12168.
Jeunemaitre X, Soubrier F, Kotelevtsev YV et al: Molecular basis of human hypertension: role of angiotensinogen. Cell 1992; 71: 169–180.
Yang W, Huang J, Ge D et al: Lipoprotein lipase gene is in linkage with blood pressure phenotypes in Chinese pedigrees. Hum Genet 2004; 115: 8–12.
Yang WJ, Huang JF, Yao CL et al: Evidence for linkage and association of the markers near the LPL gene with hypertension in Chinese families. J Med Genet 2003; 40: e57.
Tu X, Tu J, Wen X et al: A study of lipoprotein lipase gene intron 8 polymorphisms in Chinese Han race essential hypertension patients. Int J Cardiol 2005; 99: 263–267.
Hunt SC, Province MA, Atwood LD et al: No linkage of the lipoprotein lipase locus to hypertension in Caucasians. J Hypertens 1999; 17: 39–43.
Socquard E, Durlach A, Clavel C et al: Association of HindIII and PvuII genetic polymorphisms of lipoprotein lipase with lipid metabolism and macrovascular events in type 2 diabetic patients. Diabetes Metab 2006; 32: 262–269.
Zee RY, Cook NR, Cheng S et al: Polymorphism in the beta2-adrenergic receptor and lipoprotein lipase genes as risk determinants for idiopathic venous thromboembolism: a multilocus, population-based, prospective genetic analysis. Circulation 2006; 113: 2193–2200.
Bos G, Scheffer PG, Vieira D et al: The relationship of lipoprotein lipase activity and LDL size is dependent on glucose metabolism in an elderly population: the Hoorn Study. Diabetes Care 2004; 27: 796–798.
Lamarche B, Tchernof A, Moorjani S et al: Small, dense low-density lipoprotein particles as a predictor of the risk of ischemic heart disease in men: prospective results from the Quebec Cardiovascular Study. Circulation 1997; 95: 69–75.
Rip J, Nierman MC, Ross CJ et al: Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol 2006; 26: 1236–1245.
Babaev VR, Patel MB, Semenkovich CF et al: Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem 2000; 275: 26293–26299.
Mamputu JC, Levesque L, Renier G : Proliferative effect of lipoprotein lipase on human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2000; 20: 2212–2219.
Van Eck M, Zimmermann R, Groot PH et al: Role of macrophage-derived lipoprotein lipase in lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol 2000; 20: E53–E62.
Wilson K, Fry GL, Chappell DA et al: Macrophage-specific expression of human lipoprotein lipase accelerates atherosclerosis in transgenic apolipoprotein e knockout mice but not in C57BL/6 mice. Arterioscler Thromb Vasc Biol 2001; 21: 1809–1815.
Baum L, Wiebusch H, Pang CP : Role for lipoprotein lipase in Alzheimer's disease: an association study. Micros Res Tech 2000; 50: 291–296.
Fidani L, Compton D, Hardy J et al: No association between the lipoprotein lipase S447X polymorphism and Alzheimer's disease. Neurosci Lett 2002; 322: 192–194.
Blain JF, Aumont N, Théroux L et al: A polymorphism in lipoprotein lipase affects the severity of Alzheimer's disease pathophysiology. Eur J Neurosci 2006; 24: 1245–1251.
Scacchi R, Gambina G, Broggio E et al: The H+ allele of the lipoprotein lipase (LPL) HindIII intronic polymorphism and the risk for sporadic late-onset Alzheimer's disease. Neurosci Lett 2004; 367: 177–180.
Paradis E, Clement S, Julien P et al: Lipoprotein lipase affects the survival and differentiation of neural cells exposed to very low density lipoprotein. J Biol Chem 2003; 278: 9698–9705.
Paradis E, Clavel S, Julien P et al: Lipoprotein lipase and endothelial lipase expression in mouse brain: regional distribution and selective induction following kainic acid-induced lesion and focal cerebral ischemia. Neurobiol Dis 2004; 15: 312–325.
Blain JF, Paradis E, Gaudreault SB et al: A role for lipoprotein lipase during synaptic remodeling in the adult mouse brain. Neurobiol Dis 2004; 15: 510–519.
Su B, Wang X, Nunomura A et al: Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res 2008; 5: 525–532.
Le-Niculescu H, Balaraman Y, Patel S et al: Towards understanding the schizophrenia code: an expanded convergent functional genomics approach. Am J Med Genet B Neuropsychiatr Genet 2007; 144: 129–158.
Rzhetsky A, Wajngurt D, Park N et al: Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA 2007; 104: 11694–11699.
Brunner HG, van Driel MA : From syndrome families to functional genomics. Nat Rev Genet 2004; 5: 545–551.
Oti M, Huynen MA, Brunner HG : Phoneme connections. Trends Genet 2008; 24: 103–106.
Acknowledgements
We are grateful to Professor Michael Gill and Dr Derek Morris from Trinity College Dublin and anonymous peer reviewers for their valuable comments to the revision of this article. This work was partly supported by grants from Chongqing Medical University (No. 0124418029 and No. XBZD200701), the Natural Science Foundation Project of CQ CSTC (No. 2008BB5074) and The Program for Excellent Talents of University in Chongqing Municipality.
Author information
Authors and Affiliations
Corresponding author
Additional information
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Xie, C., Wang, Z., Liu, X. et al. The common biological basis for common complex diseases: evidence from lipoprotein lipase gene. Eur J Hum Genet 18, 3–7 (2010). https://doi.org/10.1038/ejhg.2009.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.134
Keywords
This article is cited by
-
Analysis of the role of Purα in the pathogenesis of Alzheimer's disease based on RNA-seq and ChIP-seq
Scientific Reports (2021)
-
HMGR overexpression and interference affect the expression of steroidogenic genes and cholesterol content in bovine intramuscular adipocytes
Scientific Reports (2020)
-
Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice
JBIC Journal of Biological Inorganic Chemistry (2020)
-
Systems Pharmacology Dissection of the Integrated Treatment for Cardiovascular and Gastrointestinal Disorders by Traditional Chinese Medicine
Scientific Reports (2016)
-
Small bowel feeding and risk of pneumonia in adult critically ill patients: a systematic review and meta-analysis of randomized trials
Critical Care (2013)
