
Abstract
Fast and efficient high-throughput techniques are essential for the molecular diagnosis of highly heterogeneous hereditary diseases, such as retinitis pigmentosa (RP). We had previously approached RP genetic testing by devising a chip based on co-segregation analysis for the autosomal recessive forms. In this study, we aimed to design a diagnostic tool for all the known genes (40 up to now) responsible for the autosomal dominant and recessive RP and Leber congenital amaurosis (LCA). This new chip analyzes 240 single nucleotide polymorphisms (SNPs) (6 per gene) on a high-throughput genotyping platform (SNPlex, Applied Biosystems), and genetic diagnosis is based on the co-segregation analysis of SNP haplotypes in independent families. In a single genotyping step, the number of RP candidates to be screened for mutations is considerably reduced, and in the most informative families, all the candidates are ruled out at once. In a panel of RP Spanish pedigrees, the disease chip became a crucial tool for selecting those suitable for genome-wide RP gene search, and saved the burdensome direct mutational screening of every known RP gene. In a large adRP family, the chip allowed ruling out of all but the causative gene, and identification of an unreported null mutation (E181X) in PRPF31. Finally, on the basis of the conservation of the SNP haplotype linked to this pathogenic variant, we propose that the E181X mutation spread through a cohort of geographically isolated families by a founder effect.
Similar content being viewed by others


Introduction
Retinitis pigmentosa (RP), the most common form of inherited retinal dystrophies (prevalence 1:3500 individuals), shows extremely high clinical and genetic heterogeneity.1, 2 To date, more than 35 genes have been associated with this disease, which follows all patterns of Mendelian inheritance: autosomal dominant – adRP, autosomal recessive – arRP, and X-linked – XLRP (Retnet, http://www.sph.uth.tmc.edu/Retnet/). Although the number of RP causative genes is continuously expanding, more than 40% of the cases remain unassigned. Direct mutational screening of all known RP genes involves the analysis of more than 500 exons and/or sequencing at least 120 kb, which is too burdensome in time and cost for most laboratories. Hence, high-throughput techniques are now being used to deal with the genetic diagnosis of heterogeneous diseases. Some commercial disease chips for retinal dystrophies (Asper Ophthalmics, Tartu, Estonia) screen for nearly all reported mutations in a cost-effective manner, although for RP, only 35% of the cases are finally diagnosed. In a previous study, we developed a novel strategy for arRP genetic diagnosis, which merged co-segregation analysis and high-throughput single nucleotide polymorphism (SNP) genotyping. This analysis allowed us to efficiently exclude non-causative RP genes (82–88%), thereby diminishing greatly the number of candidates to be sequenced per family.3
Autosomal dominant RP accounts for 20–40% of all RP cases. Eighteen causative genes (CA4, CRX, FSCN2, GUCA1B, IMPDH1, NRL, NR2E3, PRPF3, PRPF8, PRPF31, RDH12, RDS, RHO, ROM1, RP1, RP9, SEMA4A, TOPORS) have been identified so far and thus, the molecular diagnosis of adRP is also challenging. Indeed, the boundaries between genes causing dominant and recessive forms are no longer meaningful, given the increasing number of cases where the same gene explains both types of inheritance, as is the case for RHO or NRL, which were first assigned as adRP.4, 5, 6, 7, 8 The need to build a comprehensive disease chip for the autosomal RP forms also applies to the closest retinal dystrophy, Leber congenital amaurosis (LCA), which shares more than half the causative genes (seven out of thirteen) with RP (the shared genes are CRB1, CRX, IMPDH1, LRAT, RDH12, RPE65, TULP1).
Apart from genetic diagnosis, one of the key unsolved issues in RP research is to elucidate the molecular basis of the pathology as a means to design effective therapies and, to this end, the identification of novel genes is crucial. Before undertaking a genome-wide search for new causative genes, all the RP and LCA candidates have to be ruled out, and at present, indirect genetic analysis is the most suitable way to achieve this goal.
In this study, we aimed to extend the range of the chip previously designed, which now carries out a comprehensive molecular analysis of all the genes causing autosomal forms of RP and LCA.
Materials and methods
DNA from patients and families
Four adRP and seven arRP families (Figure 1) were analyzed using the RP-LCA SNP-co-segregation chip. Previous clinical examination categorized the patients as suffering from RP. Informed consent from all the family members was obtained, following the tenets of the Declaration of Helsinki. All the study concerning patient recruitment and sample collection had been approved by the Bioethics Committee of the University of Barcelona (Barcelona, Spain). DNA was obtained from blood samples using the Wizard Genomic DNA purification kit (Promega Corporation, Madison, WI, USA). DNA from 104 matched Spanish control individuals was obtained from whole blood using the method stated above.

Structure of the Spanish adRP and arRP pedigrees analyzed in this study. Seven arRP families (background in dark gray) and four adRP (background in light gray) were genotyped using the combined RP-LCA chip. Asterisks in pedigrees A6, A8 and A9 indicate probands shown in Figure 6.
Clinical examination
A large Spanish pedigree (A6) compatible with autosomal dominant RP (Figure 2) with incomplete penetrance was clinically evaluated. All affected members were diagnosed with RP after ophthalmological examination at the Hospital Universitario Central de Asturias (Oviedo, Spain). The clinical diagnosis included best-corrected visual acuity and slit lamp biomicroscopy, followed by indirect ophthalmoscopy and fundus photography after pupillary dilatation. The size and the extent of the visual-field defects were assessed with Humphrey static perimetry. Ganzfeld full-field electroretinography was performed and the electrodes were applied to the conjuntiva, following the recommendations of the IFCN committee.9 The scotopic electroretinogram (ERG) was recorded after the patient had adapted to 20 min of darkness, using a single-flash dim blue light (1 metre-candle-second) and a single-flash white light (2 metre-candle-second). The photopic ERG was recorded after 10 min adaptation to light (25 metre-candle) followed by a standard flash of 2 metre-candle-second. All ERGs were recorded in accordance with the protocol of the International Society for Clinical Electrophysiology of Vision at the Hospital San Agustín (Avilés, Spain).

Enlarged adRP pedigree A6 showing incomplete penetrance. Obligate incomplete penetrants are marked with an arrowhead. Asymptomatic incomplete penetrants revealed by the genetic analysis are indicated with an asterisk. Previously undiagnosed female V.11 (arrow) showed early symptoms of RP in a diagnostic clinical reappraisal after the genetic testing.
SNP selection
The previous autosomal recessive RP-LCA chip analyzed four SNPs for 22 arRP-arLCA genes.3 Now, the chip has been enlarged with two SNPs per gene to increase the informativity. To generate a comprehensive RP-LCA chip for the two types of autosomal forms, markers for the 14 known adRP-adLCA genes (CA4, FSCN2, GUCA1B, IMPDH1, NRL, PRPF3, PRPF8, PRPF31, RDS, ROM1, RP1, RP9, SEMA4A, TOPORS) were added, as well as for the arRP-arLCA genes, CEP290, LCA5, PRCD and RD3, all identified after the first arRP chip was designed. The six SNPs per gene were selected prioritizing the following criteria: (i) high informativity according to SNPbrowser (2007) and dbSNP (http://www.ncbi.nlm.nih.gov); (ii) position physically close to the gene, and if possible, located in the promoter, intragenic and downstream regions; (iii) that they belonged to different haplotypic blocks. The selected markers are listed in Table 1.
SNP genotyping using a high-throughput platform and haplotype analyses
Sample DNA was diluted to 20 ng/μl and a total of 1 μg per sample was arrayed in 96-well plates. SNPs were genotyped with the SNPlex (Applied Biosystems, Inc., Carlsbad, CA, USA) platform, following the instructions, protocol and software provided by the manufacturers. The platform generated raw data genotypes, which were then assigned to each individual. Haplotype and co-segregation analyses were carried out by hand.
Mutational screening of non-excluded genes
All the exons and exon–intron boundaries of non-excluded genes were subsequently directly screened for mutations. Genomic DNA of patients was amplified and sequenced with the BigDye v3.1 kit (Applied Biosystems, Inc.) in the ABI PRISM 3730 DNA sequencer (Applied Biosystems, Inc.) and compared with the wild-type gene sequence.
Reverse transcriptase–PCR analysis of PRPF31 expression
Total RNA from patients and carriers, IV.1, IV.2, IV.4, IV.5, IV.9, IV.11, of the A6 family were obtained after processing frozen white cells pelleted from 600 μl of blood, using the RiboPure-Blood purification kit (Ambion, Applied Biosystems, Carlsbad, CA, USA) and following the manufacturer's instructions. First, cDNA chains were obtained by reverse transcription with the Cells-to-cDNA kit (Ambion) with MMLV reverse transcriptase, using 0.62 μ M oligo d(T) and 1.25 μM random decamers for 15 min at 37°C plus 15 min at 39°C, and finally for 45 min at 42°C. Amplification of transcripts from PRPF31 and GAPDH (used as control) was achieved using specific primers from different exons. The PCR was carried out in a final volume of 25 μl, using the GoTaq Flexi DNA polymerase (Promega) under three different sets of conditions. For amplification of GAPDH, a two-step PCR was carried out as follows: first denaturation for 2 min at 94°C, followed by 35 cycles of 20 s at 94°C and 2 min at 63°C. For amplification of PRPF31, a three-step PCR was carried out: first denaturation for 2 min at 94°C, followed by 35 cycles of 20 s at 94°C, 30 s at 58°C and 25 s at 72°C. Longer cycles (up to 40) were carried out to detect transcripts in patients, who were heterozygotes for a null allele. The reverse transcriptase (RT)–PCR products were resolved by electrophoresis and a semiquantitative analysis was carried out using the Quantity One 4.5 software (Bio-Rad, Hercules, CA, USA). The values were normalized against GAPDH levels and represented considering the wild-type ratio PRPF31/GAPDH as 100%.
Results
Genotyping of autosomal dominant retinitis pigmentosa families
Four adRP families were analyzed by the combined RP-LCA co-segregation chip. The original chip consisted of 88 SNPs (4 SNPs for a total of 22 arRP-arLCA genes), but considering that dominant pedigrees need more genetic information for co-segregation analysis, the number of SNPs genotyped per gene was increased to six. Thus, the current chip contains 240 SNP markers covering the 40 autosomal RP/LCA genes reported at the beginning of this study.
Two of the adRP families had a large number of affected and non-affected members available for analysis (families A6 and E4, Figure 1), whereas the size of the other two was much smaller (families A8 and A9, Figure 1), with merely 2–3 affected live members. Incomplete penetrance is relatively frequent in dominantly inherited disorders, such as adRP,10, 11 and thus, it was also taken into account while carrying out the co-segregation analysis. Also in our families there were some obligate carriers, who were non-penetrant but had affected progeny. Our criterion was that all affected individuals should share one haplotype that could be present in some non-affected family members.
Overall, the efficiency of this high-throughput analysis was much higher in the two large families, as we ruled out all genes but one (maximum informativity) in family A6, and only 4 genes out of 40 (90% efficiency) remained as candidates in family E4. Even in the two small families, which were less informative because of the scarce number of meioses available, half the RP-LCA candidate genes were ruled out.
Mutational screening of the remaining candidates in the adRP families
Subsequent to the chip analysis, we aimed to identify the pathogenic mutation in those adRP families where the number of remaining candidates was manageable (<5). Thus, families A8 and A9 were not further pursued at this stage. In the two larger pedigrees, all the genes were ruled out but one in family A6 or four in family E4. In the latter, these four genes were directly sequenced but no pathogenic mutation was detected, and thus, this pedigree is now being considered for RP gene search.
Remarkably, for family A6 (Figure 2), the sequence of the single candidate left, PRPF31, revealed a new nonsense mutation, a G>T transversion (c.541 G>T) (Figure 3). This pedigree showed three asymptomatic obligate carriers (indicated with arrowheads in Figure 2), as they had several affected descendants. After haplotype analysis, four additional incomplete penetrants were identified (shown by asterisks and one arrow, Figure 2), who were later confirmed by sequence. These results prompted us to request a second clinical assessment. It must be noted that individual V.11 (aged 19), who had been previously categorized as normal was diagnosed with early symptoms of RP (arrow, Figure 2).

Identification of the c.541 G>T mutation. PRPF31 exon 6 sequence from a control and patient V.10, showing heterozygosity in position c.541(G>T).
This new mutation, c.541 G>T, introduces a nonsense codon (E181X). It has been shown that transcripts containing premature termination codons are detected by the mRNA quality control system and eliminated before translation by non-sense mediated decay (NMD).12, 13 To assess whether the E181X mutation resulted in reduced levels of PRPF31 transcripts, we carried out a semi-quantitative RT–PCR analysis of PRFP31 expression in blood white cells from a non-carrier sibling (IV.9), two full penetrants (IV.4 and IV.11) and three incomplete penetrants (IV.1, IV.2 and IV.5) (Figure 4a). Incomplete penetrants produced lower PRPF31 transcript levels than the non-carrier. Even more relevant to pathogenicity, the PRPF31 expression in full penetrants (patients) was not detected under standard RT–PCR conditions (35 cycles), but could be observed in the two patients (IV.4 and IV.11) after extending the number of PCR cycles up to 45 (Figure 4b). The relative quantitative analysis of the normalized PRPF31 mRNA expression in leukocytes after 35 cycles showed that non-penetrant heterozygotes PRPF31 levels were 51–84% of the control, in contrast to full penetrants in which PRPF31 was undetectable (Figure 4c). Taken together, our results indicate that the E181X mutation is the cause of the disorder in this family, in agreement with previous reports showing that dominant inheritance of PRPF31 mutations is mainly due to haploinsufficiency.13, 14

PRPF31 RT–PCR analysis in blood of incomplete penetrants, full penetrants and a control sibling of the A6 family. (a) Incomplete penetrants IV.1, IV.2 and IV.5 showed lower, although detectable levels of PRPF31 transcripts compared with a healthy sibling (IV.9). In contrast, PRPF31 expression was non-detected in patients IV.4 and IV.11 under standard RT–PCR conditions (35 cycles). GAPDH was used as control for normalization. (b) PRPF31 expression in patients was observed only when the number of PCR cycles was extended (up to 45). (c) Semiquantitative analysis of PRPF31 levels (at 35 cycles) using GAPDH expression for normalization, and considering the PRPF31 levels of the wild-type control (IV.9) as 100%.
Clinical assessment and findings of the A6 family
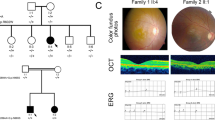
Our results highlighted several members of the family who were carriers of the E181X mutation but who had not been clinically assessed. Our genetic testing prompted a diagnostic reappraisal of the incomplete penetrants. Individuals IV.1, IV.5, IV.14 and V.11 volunteered and their phenotype were compared with that of patient V.10 (Table 2). As observed in Figure 5, patient V.10 showed conventional RP features, such as, severe night blindness, decreased visual acuity and loss of mid-peripheral visual field. In contrast, individuals IV.1, IV.5 and IV.14 presented normal ERGs and eye fundi, and hence were categorized as incomplete penetrants (Table 2 and Figures 5a–d). The loss of visual acuity in individual IV.5 was due to cataracts (Table 2), but the retina was unaffected (Figure 5b). Remarkably, the clinical evaluation of the individual V.11, who had not been previously diagnosed with RP, revealed some of the early RP traits: mild Retinal Pigmented Epithelium (RPE) atrophy in the peripheral and nasal retina, and vascular attenuation (Figure 5e). In addition, this patient showed moderate night blindness and mild visual field constriction in both eyes, the ERG showed decreased amplitude and increased latency in both rod and cone waves, and flicker response with decreased amplitude (Table 2). Therefore, in this particular case, genetic diagnosis preceded clinical phenotype and was instrumental for early detection of RP onset.

Fundus eye photographs (OS-left eye; OD-right eye) from one affected and several mutation carriers detected after genetic testing. (a) Incomplete penetrant IV.1. (b) Incomplete penetrant IV.5. (c) Incomplete penetrant IV.14. (d) Patient V.10, the view of the OD fundus is lateral to show the peripheral bone-spicule pigment deposits and vascular attenuation. (e) Previously undiagnosed female V.11, now showing early symptoms of RP, such as peripheric retinal pigmented epithelium atrophy.
Haplotype conservation as indicative of a mutational founder effect
The analysis of this large A6 pedigree allowed us to identify the haplotype linked to the new mutation. We then considered whether the pedigrees A8 and A9, which came from the same geographical area in Asturias (north west of Spain) as family A6, but with no previous record of genetic relationship, shared the causative mutation due to a founder effect. These two families were not selected earlier for mutational screening, given the high number of remaining RP candidates, among them PRPF31. Our hypothesis of common ancestry was further supported on the grounds that all affected individuals in the three families shared the SNP haplotype linked to the E181X mutation (Figure 6). Direct sequencing revealed that the RP patients in the A8 and A9 families carried the same mutation.

Single nucleotide polymorphism (SNP) haplotype conservation in patients from A6, A8 and A9 families. The PRPF31 SNP haplotype co-segregating with the disease is conserved in three families from the same geographical region, as exemplified by one affected member of each family (gray bar). The relative location with respect PRPF31 (black box) of the SNPs and their chromosomal position is also depicted. The SNP genotypes are given by the SNPlex design: the first five correspond to the allele of the plus chromosomal strand, whereas the last one is read on the minus strand.
We believe that these results provide a proof-of-principle for identifying mutational founder effects, and underscore the usefulness of haplotype co-segregation analysis based on SNPs located close to RP genes, particularly in families with poor informativity.
Genotyping and mutational screening of autosomal recessive retinitis pigmentosa families
Seven arRP families were also collected (Figure 1) and analyzed by the RP-LCA co-segregation chip. All of them were consanguineous and thus, RP candidates must abide to both, co-segregation and homozygosity-by-descent criteria. The chip efficiency in these cases was extremely high (95%), as an average of only two genes remained as candidates per family. In two large pedigrees (A1 and A5, Figure 1), all the RP-LCA genes (dominant and recessive) were directly ruled out after this single genotyping step. Hence, we are now undertaking genome-wide gene identification in these two families. Concerning the five remaining arRP pedigrees, the non-excluded candidates were screened for mutational analysis but no pathogenic mutation was found. However, their small size did not warrant statistical significance in linkage analysis.
Discussion
As more causative genes are identified in highly heterogeneous diseases, the more imperative is the use of a comprehensive high-throughput approach for molecular diagnoses. Our SNP-based co-segregation chip for RP and LCA genetic testing is fast, reliable and cost-effective, as it directly rules out a high number of retinal dystrophy candidates and highlights those for mutational screening. From the technical point of view, our multiplex strategy is very flexible, as additional SNPs for new genes can be easily incorporated. Moreover, no previous knowledge of reported mutations is required, as the main criterion for candidate inclusion is co-segregation with the disease. The key issue of this type of analysis is genetic informativity, which depends on the average heterozygosity of the genetic markers and the number of meioses available. The selection of SNPs is of utmost relevance, as they have to be both very informative and close enough to the gene to minimize double recombination events. To increase the exclusion score of our RP-LCA chip, the number of SNPs per gene has been raised from four to six. As a result, the number of candidates ruled out per family has increased, reducing the number of genes left for mutational screening. In fact, although the number of recessive consanguineous families used in this study did not allow for statistical significance, the comparison of the same genes analyzed in the first chip versus this new comprehensive version showed that the average of non-excluded arRP candidates per family dropped from 2.7 to 1.
The efficiency of the genetic analysis depends on the structure of the pedigrees and the type of inheritance. Therefore, dominant forms – where only one allele causes the disease – require a higher number of affected individuals compared with recessive forms, in which all the affected siblings have to share the two alleles. Furthermore, in consanguineous cases homozygosity-by-descent adds further constraint to mere co-segregation, making the molecular diagnosis of even a single-affected family member possible. According to our results, dominant pedigrees with a minimum of four to five affected individuals are informative enough to be effectively screened by our chip. In this context, family A6 was paradigmatic, as all candidates but the causative gene were directly excluded. Mutational screening of this family by direct sequencing allowed us to identify a new nonsense mutation in position c.541 G>T of PRPF31, which introduced a premature termination codon (E181X). Interestingly, PRPF31 is ubiquitiously expressed as it encodes a pre-mRNA splicing factor, which is an integral factor of the snRNP complex (spliceosome). Yet, the only phenotype associated to PRPF31 mutations is RP. It can be noted that, other pre-mRNA splicing factors, PRPF3 and PRFP8, are also involved in autosomal dominant RP. Many PRPF31 pathogenic alleles are null mutations, which have been previously shown to cause RP by haploinsufficiency, mainly through the NMD surveillance mechanism.10, 13, 14 The variation in the levels of expression of the remaining wild-type allele directly correlates with incomplete penetrance in pedigrees.11 Moreover, this expression variation has been shown to be a highly heritable character, depending on at least two transacting expression quantitative trait loci (eQTLs) (expression quantitative trait locus), which would therefore act as genetic modifiers.15 In our case, the mRNA levels in the blood of symptomatic and asymptomatic individuals support the pathogenicity of the identified mutation in the A6 family, and confirm that the disease depends on the amount of wild-type mRNA produced.
This disease chip requires familial cases for co-segregation analysis, and although family sample collection is more demanding than the mere analysis of probands, the benefits derived from this additional information are worthwhile as: (i) it directly detects presymptomatic carriers (as in the case of individual V.11 in pedigree A6); (ii) it facilitates prenatal diagnosis and genetic counselling; and (iii) it identifies mutation-associated SNP haplotypes, which under the assumption of a mutational founder effect, provide the genetic basis of the disease in families otherwise unsuitable for linkage analysis.
Finally, our approach highlights the families in which all the known candidate genes are excluded, thus singling out the pedigrees for the identification of novel RP genes. As 40% of the RP cases remain unassigned, this still is one of the main challenges in the inherited retinal disorders.
Conflict of interest
The authors declare no conflict of interest.
References
Rivolta C, Sharon D, DeAngelis MM, Dryja TP : Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet 2002; 11: 1219–1227.
Hims MM, Diager SP, Inglehearn CF : Retinitis pigmentosa: genes, proteins and prospects. Dev Ophthalmol 2003; 37: 109–125.
Pomares E, Marfany G, Brion MJ, Carracedo A, Gonzalez-Duarte R : Novel high-throughput SNP genotyping cosegregation analysis for genetic diagnosis of autosomal recessive retinitis pigmentosa and Leber congenital amaurosis. Hum Mutat 2007; 28: 511–516.
Dryja TP, McGee TL, Reichel E et al: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990; 343: 364–366.
Farrar GJ, McWilliam P, Bradley DG et al: Autosomal dominant retinitis pigmentosa: Linkage to rhodopsin and evidence for genetic heterogeneity. Genomics 1990; 8: 35–40.
Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP : A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet 1992; 1: 209–213.
Bessant DA, Payne AM, Mitton KP et al: A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet 1999; 21: 355–356.
Nishiguchi KM, Friedman JS, Sandberg MA, Swaroop A, Berson EL, Dryja TP : Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci USA 2004; 101: 17819–17824.
Celesia GG, Bodis-Wollner I, Chatrian GE, Harding GF, Sokol S, Spekreijse H : Recommended standards for electroretinograms and visual evoked potentials. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87: 421–436.
Vithana EN, Abu-Safieh L, Pelosini L et al: Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci 2003; 44: 4204–4209.
Sato M, Nakazawa M, Usui T, Tanimoto N, Abe H, Ohguro H : Mutations in the gene coding for guanylate cyclase-activating protein 2 (GUCA1B gene) in patients with autosomal dominant retinal dystrophies. Graefes Arch Clin Exp Ophthalmol 2005; 243: 235–242.
Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP : Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat 2006; 27: 644–653.
Rio Frio T, Wade NM, Ransijn A, Berson EL, Beckmann JS, Rivolta C : Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J Clin Invest 2008; 118: 1519–1531.
Abu-Safieh L, Vithana EN, Mantel I et al: A large deletion in the adRP gene PRPF31: evidence that haploinsufficiency is the cause of disease. Mol Vis 2006; 12: 384–388.
Rio Frio T, Civic N, Ransijn A, Beckmann JS, Rivolta C : Two trans-acting eQTLs modulate the penetrance of PRPF31 mutations. Hum Mol Genet 2008; 17: 3154–3165.
Acknowledgements
We are grateful to the members of all the families for their participation in this study. We are obliged to A Mayor for sample collection, helpful discussions and constant support to our research. EP was a recipient of an FPI fellowship from the Spanish Ministry of Education and Science (MEC), and is now under contract by CIBERER. MR was a recipient of a scholarship from Bidons Egara and is now a recipient of an FPU fellowship from MEC. PM and JP are under contract by CIBERER. This study was funded by Fundaluce (2004), Grant BFU2006-04562 (Ministerio de Educación y Ciencia) and CIBERER INTRA/07–08/718.1 to RG-D. This study was approved by the Bioethics Committee of the University of Barcelona (Barcelona, Spain).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pomares, E., Riera, M., Permanyer, J. et al. Comprehensive SNP-chip for retinitis pigmentosa-Leber congenital amaurosis diagnosis: new mutations and detection of mutational founder effects. Eur J Hum Genet 18, 118–124 (2010). https://doi.org/10.1038/ejhg.2009.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.114
Keywords
This article is cited by
-
Molecular characterization of AIPL1 gene region in the Iranian population: application of novel informative haplotypes and detection of mutational founder effect
Genes & Genomics (2017)
-
SAQC: SNP Array Quality Control
BMC Bioinformatics (2011)
