
Abstract
In most Dutch melanoma families, a founder deletion in the melanoma susceptibility gene CDKN2A (which encodes p16) is present. This founder deletion (p16-Leiden) accounts for a significant proportion of the increased melanoma risk. However, it does not account for the Atypical Nevus (AN) phenotype that segregates in both p16-Leiden carriers and non-carriers. The AN-affected p16-Leiden family members are therefore a unique valuable resource for unraveling the genetic etiology of the AN phenotype, which is considered both a risk factor and a precursor lesion for melanoma. In this study, we performed a genome-wide scan for linkage in four p16-Leiden melanoma pedigrees, classifying family members with five or more AN as affected. The strongest evidence for an atypical nevus susceptibility gene was mapped to chromosome band 7q21.3 (two-point LOD score=2.751), a region containing candidate gene CDK6.
Similar content being viewed by others
Introduction
Approximately 10% of melanoma cases are thought to be familial.1 Familial melanoma is defined by the occurrence of melanoma in at least two first-degree relatives. Familial melanoma and Atypical Nevi (AN) were originally thought to be pleiotropic effects of the same gene. Familial melanoma is therefore often referred to as Familial Atypical Multiple Mole Melanoma (FAMMM) syndrome.2 A mutation is found in approximately 40% of melanoma families (with at least three melanoma patients) in the cell-cycle regulator CDKN2A. CDKN2A maps to chromosome band 9p21 and encodes the proteins p16 and p14ARF.3, 4, 5 However, increasing evidence has emerged against atypical nevi being caused by the CDKN2A melanoma susceptibility gene alone.6, 7, 8
Melanoma families have been ascertained in the Leiden University Medical Center since the early 1980s. These families have been part of an ongoing effort to gain insight into the etiology of melanoma. The families originate from two towns that are known for their isolated character and their tight family connections. Identification of haplotype sharing across p16-Leiden families, together with a genealogical evaluation of this relatively isolated population, suggests sharing of a common founder in these families.9
Linkage analysis performed in 1993 on the Leiden FAMMM family members provided evidence against linkage of the AN phenotype to the 9p melanoma susceptibility locus. Considering the six families from the same region, inclusion of family members with 10 or more AN as affected led to a slight decrease in LOD score from 2.78 (θ=0.00) to 2.64 (θ=0.08) for marker D9S171. Inclusion of persons with five or less AN as affected resulted in a remarkably decreased LOD score of 1.52 (θ=0.13), suggesting that the presence of AN could not entirely be ascribed to the melanoma susceptibility gene.10 Identification of the p16-Leiden founder mutation (a 19-bp deletion) in these families highlighted the complexity of the FAMMM phenotype and its underlying causes. For instance, several family members with a florid manifestation of the AN phenotype, even some who developed melanoma, did not carry the p16-Leiden deletion.11 This finding has also been reported by other authors,6, 12 suggesting that other factors, either environmental or genetic, contribute to AN development.
Now, 10 years later, intense follow-up of our large extended founder pedigrees with many AN-affected family members in combination with advanced technology offers a unique opportunity for investigating the genetic component of AN.
Subjects and methods
Study inclusion
Melanoma families were ascertained through the Pigmented Lesions Clinic of the Leiden University Medical Center since 1980, and were included in a registry. As the genotype status was unknown at that time, the inclusion of family members was independent of the p16-Leiden status. For this study, members of p16-Leiden mutation-positive families were asked to participate via a letter from the principal investigator. The participating family members were seen by a research nurse and a dermatologist for thorough investigation of their nevi at the Pigmented Lesions Clinic. The family members were examined prior to the determination of carrier status. After informed consent of the family members, blood was drawn for DNA isolation. The p16-Leiden carrier status was determined using PCR technology as previously described.13
Description of phenotype
A nevus was considered atypical when it had a flat component and fulfilled three or more of the following criteria: size of 5 mm or larger, vague border, red hue, asymmetrical outline, variable pigmentation. As the true phenotype of the suspected ‘atypical nevus gene’ is not yet known, we have decided to consider the threshold of having five or more AN as being affected, this being a relatively rare phenotype of 6% in the general population (unpublished results, W Bergman).
Genotyping
A genome-wide scan was performed using 414 microsatellite markers with an average of 10 cM between markers and with approximately 80% heterozygosity. A TECAN Genesis 200 robot was used to set up the PCRs with fluorescent primers. The PCR conditions were as follows: 20 ng of DNA, 0.33 μ M of each primer, 0.25 mM of each dNTP, 2.5 mM MgCl2, 10 mM Tris-HCl, 50 mM KCl and 0.5 U Taq polymerase. PCR cycling conditions were as follows: 95°C for 12 min, followed by 10 cycles of 94°C for 15 s, 55°C for 15 s, 72°C for 30 s, and 20 cycles of 89°C for 15 s, 55°C for 15 s, and 72°C for 30 s, with a final extension of 72°C for 10 min. The PCR products were separated using the ABI 3100 DNA sequencer. The ROX 400 size standard was run as an internal size standard. Allele sizing and labeling were performed using GENESCAN and GENOTYPER software (Applied Biosystems) by two persons separately for quality control purposes. A CEPH control individual and 10 encoded duplicate samples were included for quality control purposes.
Statistical analyses
Prior to the linkage analysis, family structure and biological kinships were examined by comparing the expected and observed average identity-by-state of genotyped markers in all pairs of individuals using the Graphical Relationship Representation program.14 Genotyping was then checked for Mendelian inconsistencies using the Pedstats program. In the rare case of genotypes violating Mendelian transmission, they were re-analyzed by checking the allele calling in GENOTYPER. If ambiguity remained, the marker genotypes were excluded from further analyses. Marker order and distances (Haldane cM) were interpolated via locally weighted linear regression from NCBI build 35.1 physical map positions and the Rutgers genetic map.15, 16 Allele frequencies were estimated from all genotyped individuals with the MERLIN program. For every marker, two-point non-parametric and parametric linkage analyses were performed using MERLIN.17 The parametric analysis was done under a dominant model with a disease allele frequency of 0.025, a phenocopy rate of 3% and a penetrance of 60% in gene carriers. Individuals with five or more AN were coded as affected, whereas all others were coded as unknown. Linkage findings were evaluated with simulation studies using the gene-drop option of MERLIN. One thousand genome-wide scan replicates were generated based on the existing marker allele frequencies and analyzed as the real data. The empirical LOD score distributions obtained from these simulations were used to determine the significance of the observed LOD scores. Evidence for linkage was evaluated using the criteria of Lander and Kruglyak with empirical thresholds for suggestive linkage, where one marker in every genome scan is false positive, and for significant linkage, where one in every 20 genome scans is false positive.18 For the parametric linkage analysis, suggestive linkage is obtained at LOD≥1.99 and significant linkage at LOD≥2.98. For the non-parametric linkage analysis, these values were LOD≥0.99 and LOD≥1.02. All LOD scores greater than 0.99 were reported.
After additional markers were genotyped in the most promising regions, multipoint non-parametric and parametric linkage analyses were performed with the MERLIN program. For the parametric analysis, we employed the same model that was used in the single-point analysis. For these analyses, the LOD scores and asymptotic level of significance from MERLIN were reported as empirical P-values would take too much time without the limitation of a maximum of two recombinations that were used to enable analysis of the large families.
Results
In the four participating p16-Leiden families, there were 21 individuals affected with AN and negative for the p16-Leiden mutation. The total number of family members included in our analysis is 113 individuals (Figure 1) including 88 with DNA available (21 affected and 67 unknown).

Pedigrees of the four included families 19, 3.1, 3.3 and 10.2. ○=female family member; □=male family member; ▪ or •=AN affected; •=genotyped. The number underneath the 21 affected family members indicates the number of atypical nevi exhibited by this family member. Family members without nevus counts are considered unknown. The total number of family members included in our analysis is 113 individuals, including 88 with DNA available (21 affected and 67 unknown).
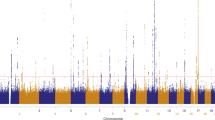
For the genome-wide scan with 409 markers (414 markers minus ambiguous markers), we had a genotyping success rate of 94%. Only a single difference was found out of 3655 duplicated genotypes, leading to an error rate of 0.027%. The parametric and non-parametric results of the genome-wide scan for all families combined are shown in Figure 2.

Genome-wide two-point linkage results for 409 markers for families 19, 3.1, 3.3 and 10.2 combined. The position of the chromosomes is provided.
Parametric linkage: marker D7S657 showed suggestive evidence of linkage, with the highest LOD score of 2.75 at position 104.9 cM on chromosome 7 (7q21.3). The genome-wide significance level of this marker was P=0.000106. The flanking marker D7S515 yielded an LOD score of 1.24. In addition, we observed LOD scores greater than 1 on chromosomes 2p12, 4q13.3, 6p22.3, 8q22.3 and 9q22.2 (Table 1).
Non-parametric linkage: overall non-parametric results were consistent with parametric results. Simulation results for the calculation of empirical P-values indicated that an LOD score of 0.99 determines suggestive linkage and LOD=1.02 determines significant linkage. In Table 1, the results from the non-parametric analysis thus show two additional markers with linkage evidence, namely D9S1338 (LOD=1.16) and D12S86 (LOD=1.04). Significant linkage was also found for markers D4S392 (LOD=1.32) and D7S657 (LOD=1.36), previously identified with the parametric linkage analysis. For the remaining markers identified with the parametric linkage analysis, the non-parametric LOD scores were below 0.99. For all markers in Table 1, empirical P-values were below 0.05, with the exception of the parametric results for D9S1338 and D12S86.
Given the maximum resulting LOD score of 2.75, the evidence for linkage using two analysis methods and a positive flanking marker D7S515, we subsequently genotyped six additional markers (D7S492, D7S2410, D7S2409, D7S646, D7S2431 and UT682) in the 7q21.3 region (Table 1). The results of the parametric and non-parametric multipoint analyses for these markers are shown in Figure 3. An LOD score of 2.05 in the multipoint analysis was found at marker D7S2410 (P=0.002). For the non-parametric analysis, the highest LOD score at marker D7S2410 was 1.17 (P=0.01).

Multipoint LOD scores for families 19, 3.1, 3.3 and 10.2 for nine markers analyzed on chromosome 7. The position of the markers is indicated by triangles. The marker order and recombination distances are according to the recent physical map (Ensembl.org) and integrated Rutgers map. Markers indicated with an asterisk are markers from the original screen.
Haplotype construction for eight chromosome-7 markers is shown in Figures 4a–d for the affected individuals. This construction shows no linkage in the uninformative family 10.2, and partial sharing in both families 3.3 and 3.1. Family 19 also shows linkage; however, there is one substantial branch without sharing the linked haplotype. Between families, a shared haplotype is not present.

(a–d) Haplotype construction for the eight chromosome-7 markers for the affected individuals per family. Markers with an asterisk are markers from the original screen.
Discussion
Since the discovery of CDKN2A as an important melanoma susceptibility gene, there has been a remarkable increase in our knowledge of genotype–phenotype correlations between mutations in this gene and melanoma as well as other tumors.19, 20 Despite this increasing knowledge, there still remains a great gap in understanding the nevus phenotype often seen in the FAMMM syndrome. Although the melanoma susceptibility genes may be nevogenic to a certain extent, the underlying cause of most atypical nevi still lacks a satisfactory explanation.6, 11, 12
We have undertaken a genome-wide scan for linkage in four p16-Leiden melanoma pedigrees to localize novel atypical nevus susceptibility genes. The 7q21.3 region showed the strongest evidence for linkage: marker D7S657 had a two-point LOD score of 2.75 and D7S515, which flanks D7S657, had an LOD score of 1.24 (empirical P-values). Non-parametric results showed a similar finding with an LOD score of 1.36 for marker D7S657, and also an empirical P-value below 0.001.This prompted us to further investigate this region as a novel atypical nevus locus. Follow-up genotyping did not increase the evidence for linkage in this region. Haplotype analysis indicated this region not being a shared haplotype. We did proceed with sequence analysis of one candidate gene, CDK6, located in this region. This gene plays a critical role in the cell-cycle regulation pathway: Together with CDK4, CDK6 is directly inhibited by P16. However, no pathogenic mutations were found in its sequence. No further investigation such as expression studies has been performed so far to study the possible involvement of this gene in AN development.
Finding underlying causes for complex genetic disorders is a difficult task. In an effort to minimize heterogeneity and increase the power of this study, we used family members from a proven founder population. A larger number of affected individuals would in this case further increase the power of detecting a nevus susceptibility locus.
We have analyzed our data using both parametric and non-parametric analyses to limit the disadvantage of misspecification of our disease model in the parametric analysis and to compare results. Generally, the resolution and LOD scores of the non-parametric model were lower. This was either a result of a smaller detection power in the non-parametric analysis, or by overestimation of the contribution of the AN gene in our parametric model. The single-point empirical P-values obtained with the two methods were very similar despite large LOD score differences between the analysis methods.
We should consider that the lack of an accepted and reproducible definition of the nevus phenotype21 further complicates this study, potentially leading to biased results. Ideally, one would have liked to analyze this phenotype as a quantitative trait; however, atypical nevus count is a truncated, not normal, distribution and the family members included in this analysis are highly ascertained, making a quantitative trait analysis practically impossible.
Although ample evidence has demonstrated the heritability of AN,22, 23, 24 uncertainty regarding the ‘true’ phenotype will only be resolved with more knowledge of AN pathogenesis. To minimize the misdiagnoses of the phenotype in this study, we have used a rather strict definition (five or more AN). Exploratory analyses could help to determine how sensitive our results are to this threshold. The nevus count was not considered as a phenotype for linkage analysis, as the distribution of counts was severely skewed within these families.
Only recently, two papers have been published on genome-wide scans for nevi: Falchi et al25 have undertaken an analysis of total nevus count in 194 twin families and Zhu et al26 report genome-wide scans for flat, raised and atypical nevi in 424 twin families. Although each of these studies have used different phenotype definitions, it is difficult to overlook a shared increased LOD score on chromosome 9q; we report an LOD score of 1.49 for marker D9S283 on 9q22, for which Zhu et al26 report an LOD score of 1.78. Falchi et al25 report an LOD score of 2.55 on marker D9S167, which is more proximal on 9q21. A collaborative effort using dense markers on shared peaks to investigate the value of these findings might be tempting; however, the enormous variability in study design might hamper this approach. A collaborative effort with a shared phenotype definition in a large population seems a reasonable approach to try to validate these findings.
In summary, we have found unconfirmed indication for an atypical nevus susceptibility gene on chromosome band 7q21.3 through a genome-wide scan for linkage in four p16-Leiden melanoma pedigrees.
References
Skolnick MH, Cannon-Albright LA, Kamb A : Genetic predisposition to melanoma. Eur J Cancer 1994; 30A: 1991–1995.
Lynch HT, Frichot III BC, Lynch JF : Familial atypical multiple mole-melanoma syndrome. J Med Genet 1978; 15: 352–356.
Kamb A, Gruis NA, Weaver-Feldhaus J et al: A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994; 264: 436–440.
Foulkes WD, Flanders TY, Pollock PM, Hayward NK : The CDKN2A (p16) gene and human cancer. Mol Med 1997; 3: 5–20.
Kefford R, Bishop JN, Tucker M et al: Genetic testing for melanoma. Lancet Oncol 2002; 3: 653–654.
Hussussian CJ, Struewing JP, Goldstein AM et al: Germline p16 mutations in familial melanoma. Nat Genet 1994; 8: 5–21.
Cannon-Albright LA, Meyer LJ, Goldgar DE et al: Penetrance and expressivity of the chromosome 9p melanoma susceptibility locus (MLM). Cancer Res 1994; 54: 6041–6044.
Bishop JA, Wachsmuth RC, Harland M et al: Genotype/phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J Invest Dermatol 2000; 114: 28–33.
Hille ET, van Duijn E, Gruis NA, Rosendaal FR, Bergman w, Vandenbroucke JP : Excess cancer mortality in six Dutch pedigrees with the familial atypical multiple mole-melanoma syndrome from 1830 to 1994. J Invest Dermatol 1998; 110: 788–792.
Gruis NA, Sandkuijl LA, Weber JL et al: Linkage analysis in Dutch familial atypical multiple mole-melanoma (FAMMM) syndrome families. Effect of naevus count. Melanoma Res 1993; 3: 271–277.
Gruis NA, Sandkuijl LA, van der Velden PA, Bergman W, Frants RR : CDKN2 explains part of the clinical phenotype in Dutch familial atypical multiple-mole melanoma (FAMMM) syndrome families. Melanoma Res 1995; 5: 169–177.
Puig S, Ruiz A, Castel T et al: Inherited susceptibility to several cancers but absence of linkage between dysplastic nevus syndrome and CDKN2A in a melanoma family with a mutation in the CDKN2A (P16INK4A) gene. Hum Genet 1997; 101: 359–364.
Gruis NA, van der Velden PA, Sandkuijl LA et al: Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat Genet 1995; 10: 351–353.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : GRR: graphical representation of relationship errors. Bioinformatics 2001; 17: 742–743.
Duffy DL : An integrated genetic map for linkage analysis. Behav Genet 2006; 36: 4–6.
Kong X, Murphy K, Raj T, He C, White PS, Matise TC : A combined linkage-physical map of the human genome. Am J Hum Genet 2004; 75: 1143–1148.
Lathrop GM, Lalouel JM : Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 1984; 36: 460–465.
Lander E, Kruglyak L : Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 1995; 11: 241–247.
Bishop DT, Demenais F, Goldstein AM et al: Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 2002; 94: 894–903.
Goldstein AM, Struewing JP, Fraser MC, Smith MW, Tucker MA : Prospective risk of cancer in CDKN2A germline mutation carriers. J Med Genet 2004; 41: 421–424.
Cramer SF : Atypical histologic features in melanocytic nevi. Am J Dermatopathol 2001; 23: 160–161.
Easton DF, Cox GM, Macdonald AM, Ponder BA : Genetic susceptibility to naevi – a twin study. Br J Cancer 1991; 64: 1164–1167.
Wachsmuth RC, Gaut RM, Barrett JH et al: Heritability and gene-environment interactions for melanocytic nevus density examined in a UK adolescent twin study. J Invest Dermatol 2001; 117: 348–352.
Goldstein AM, Tucker MA, Crutcher WA, Hartge P, Sagebiel RW : The inheritance pattern of dysplastic naevi in families of dysplastic naevus patients. Melanoma Res 1993; 3: 15–22.
Falchi M, Spector TD, Perks U, Kato BS, Bataille V : Genome-wide search for nevus density shows linkage to two melanoma loci on chromosome 9 and identifies a new QTL on 5q31 in an adult twin cohort. Hum Mol Genet 2006; 15: 2975–2979.
Zhu G, Montgomery GW, James MR et al: A genome-wide scan for naevus count: linkage to CDKN2A and to other chromosome regions. Eur J Hum Genet 2007; 15: 94–102.
Acknowledgements
We are indebted to the family members for their past and recent participation in our research. FAS is supported by the Dutch Cancer Society (RUL 99-1932); NAG is a recipient of an Aspasia fellowship of the Netherlands organization for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Snoo, F., Hottenga, JJ., Gillanders, E. et al. Genome-wide linkage scan for atypical nevi in p16-Leiden melanoma families. Eur J Hum Genet 16, 1135–1141 (2008). https://doi.org/10.1038/ejhg.2008.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.72
Keywords
This article is cited by
-
Melanoma susceptibility as a complex trait: genetic variation controls all stages of tumor progression
Oncogene (2015)
-
Association of Genetic Variants in CDK6 and XRCC1 with the Risk of Dysplastic Nevi in Melanoma-Prone Families
Journal of Investigative Dermatology (2014)
