
Abstract
Mutations in mitochondrial tRNA (mt-tRNA) genes are well recognized as a common cause of human disease, exhibiting a significant degree of clinical heterogeneity. While these differences are explicable, in part, by differences in the innate pathogenicity of the mutation, its distribution and abundance, other factors, including nuclear genetic background, mitochondrial DNA (mtDNA) haplotype and additional mtDNA mutations may influence the expression of mt-tRNA mutations. We describe the clinical, biochemical and molecular findings in a family with progressive myopathy, deafness and diabetes and striking respiratory chain abnormalities due to a well-characterized heteroplasmic mt-tRNA mutation in the mt-tRNASer(UCN) (MTTS1) gene. In addition to the m.7472Cins mutation, all individuals were homoplasmic for another variant, m.7472A>C, affecting the adjacent nucleotide in the mt-tRNASer(UCN) structure. In addition to available patient tissues, we have analysed transmitochondrial cybrid clones harbouring homoplasmic levels of m.7472A>C and varying levels of the m.7472Cins mutation in an attempt to clarify the precise role of the m.7472A>C transversion in the underlying respiratory chain abnormality. Evidence from both in vivo and in vitro studies demonstrate that the m.7472A>C is able to modify the expression of the m.7472Cins mutation and would suggest that it is not a neutral variant but appears to cause a biochemical defect by itself, confirming that homoplasmic mtDNA variants can modulate the phenotypic expression of pathogenic, heteroplasmic mtDNA mutations.
Similar content being viewed by others


Introduction
An extensive range of molecular defects has been identified in the human mitochondrial genome (mtDNA), leading to disease.1, 2 The clinical phenotypes attributable to these mutations are remarkably variable and considerable overlap exists. While these differences in phenotype and severity are explicable, in part, by variation in the distribution and abundance of the mutation, other factors, including innate pathogenicity of specific mutations, may be involved. In addition, the expression of pathogenic mtDNA mutations may be influenced by moderating genetic factors including nuclear background, mitochondrial haplotype and additional modulating mutations in the mitochondrial genome.3, 4
Pathogenic point mutations affecting mitochondrial tRNA (mt-tRNA) genes are widely described. At least eight point mutations have been previously described in the mt-tRNASer(UCN) (MTTS1) gene, many of which are associated with a mild phenotype consisting predominantly or exclusively of deafness.5, 6, 7 This is the case for many patients that harbour the well-characterized m.7472Cins MTTS1 mutation,8 though in some families, mutant loads of >95% can lead to other neurological features including myoclonus and ataxia.9, 10 The m.7472Cins mutation was first documented in a large Sicilian family, with pathogenicity confirmed in transmitochondrial cybrids which showed a decreased complex I activity and low oxygen consumption, indicative of a respiratory defect.9 Several other cases of this mutation have since been described, and cybrids harbouring the m.7472Cins have been used to explore the underlying molecular mechanisms.11, 12 Cybrid cells homoplasmic for the m.7472Cins mutation exhibited a ∼65% decrease in steady-state mt-tRNASer(UCN) levels, but with only a mild decrease in mitochondrial protein synthesis;11 a level of ∼40% mt-tRNASer(UCN) is proposed to be required for a normal rate of translation to occur.13 Further analysis revealed that the m.7472Cins mutation had no effect on the structure or rate of turnover of mt-tRNASer(UCN) but was associated with a decrease in aminoacylation by ∼25%,12 suggesting that the main mechanism of pathogenicity is the impairment of synthesis of the mt-tRNA, by both 5′ and 3′ misprocessing.14
Two recent reports have documented patients who have the m.7472Cins mutation alongside a second mutation, an A>C transversion at the same position in the mitochondrial genome.15, 16 Distribution of the m.7472A>C did not correlate with the level of m.7472Cins heteroplasmy in the family with rapid neurodegeneration described by Cardaioli et al, suggesting that the segregation of the two mutant genome types is independent. The m.7472A>C mutation however has never been documented alone, and while these papers propose the m.7472A>C to be polymorphic, they infer that it may act to suppress the pathogenic phenotype of the m.7472Cins mutation.
Here, we describe three maternally related members of a family presenting with progressive limb-girdle muscular weakness, diabetes and deafness, who have both the m.7472Cins and m.7472A>C MTTS1 gene mutations. In order to assess the influence of this second variant on the expression of the well-characterized m.7472Cins mutation, we have investigated the biochemical and molecular consequences of the m.7472Cins and the m.7472A>C in patient tissues harbouring different levels of the m.7472Cins mutation, and in transmitochondrial cybrid clones harbouring either both the m.7472Cins and the m.7472A>C mutations, or the m.7472A>C mutation alone. Our data convincingly show that cells containing only the m.7472A>C mutation exhibit a respiratory deficient phenotype, and as such this mutation might also be considered pathogenic.
Patient and methods
Case report
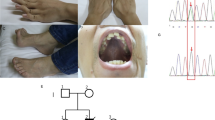
The proband (patient III-7, Figure 1) was a 53-year-old North American Caucasian woman with short stature (151 cm), migraine headaches, a long history of fatigability and a 10-year history of progressive, severe predominantly proximal weakness associated in the past year with hearing loss. Resting blood lactate and creatine kinase (CK) were normal. Cycle exercise testing performed at age 46 revealed low oxidative capacity (peak oxygen utilization 11 ml/kg per min, healthy aged match females ∼27 ml/kg per min), high exercise cardiac output relative to oxygen utilization and low extraction of available oxygen from blood characteristic of severe muscle oxidative defects.17

Pedigree highlighting the variable clinical presentation in the family and the level of the m.7472Cins mutation in family members available for testing. The proband is shown by an arrow.
Her 50-year-old brother (patient III-6) was never athletic and fatigued easily, compared to his peers as a child. In his mid-30s, he began experiencing progressive leg weakness and by his late 30s developed diabetes. By age 45, he walked with difficulty and used a motorized scooter; after sustaining a fractured humerus in a fall, he became bed bound. In his 40s, he developed progressive hearing loss. In the last 6 months of life, he developed refractory congestive heart failure complicated by aspiration pneumonia and sustained a respiratory arrest. During a clinic visit 18 months before his death, he was confined to a wheelchair with marked proximal hip and shoulder-girdle weakness. Resting blood lactate was 2.7 mM (normal, ≤1.5 mM); CK was 80 (normal, ≤269). Several tissues were obtained post-mortem from patient III-6 for analysis.
Their 72-year-old mother (II-5) had short stature (151 cm), a 20-year-history of progressive proximal weakness and deafness. Resting blood lactate was 2 mM, serum CK was normal. This patient's mother (I-1, deceased at age 93) and two older sisters aged 89 (II-1) and 89 (II-2) had deafness without weakness. The 50-year-old daughter (III-5) of II-2 was asymptomatic. Other family members were unavailable for clinical evaluation or genetic testing (Figure 1).
Muscle histochemistry and biochemistry
Histological and histochemical analyses were performed using standard procedures.18 The activities of the individual respiratory chain complexes were measured in post 600gav muscle supernatants and expressed relative to the activity of the matrix enzyme citrate synthase.19
Generation of transmitochondrial cybrid clones
Transmitochondrial cybrid clones were generated by fusion of enucleated patient platelets with ρ°cells (143BTK−ρ° osteosarcoma cell line).20 Selected cybrid clones and the 143B parental and 143B ρ° cell lines were grown in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% FCS, 110 μg/ml sodium pyruvate and 50 μg/ml uridine.
Mitochondrial DNA analyses
Total DNA was extracted from patient tissues and from individual muscle fibres.18 The entire coding region of the mitochondrial genome was amplified using a series of M13-tailed oligonucleotide primer pairs21 and compared to the revised Cambridge reference sequence, while the presence of the m.7472Cins and the m.7472A>C transversion mutations were further investigated by PCR-RFLP analysis as follows:
m.7472Cins mutation
A 208 bp fragment encompassing the mutation site in MTTS1 was amplified using primers previously described.9 The m.7472Cins mutation creates a site for the restriction endonuclease XcmI, cleaving the 208 bp fragment into two smaller products of 168 and 40 bp. Total DNA from a transmitochondrial cybrid clone harbouring homoplasmic levels of the m.7472Cins mutation alone (kindly provided by Professor Howard Jacobs, University of Tampere, Finland) was included in analyses as a control to ensure complete enzyme digestion.
m.7472A>C transversion
A reverse mismatch primer was employed to introduce a recognition site for the restriction endonuclease StyI in the presence of the mutation. Oligonucleotide primers recognized the following sequences: Light strand (7321–7341) GAGAAGCCTTCGCTTCGAAGC and heavy strand (7500–7473) GGCCATGGGGTTGGCTTGAAACCACCTT (mismatch nucleotide shown in bold). The mismatch in the antisense primer creates a StyI restriction site in the mutant PCR product which cuts the 180 bp amplimer into two fragments of 152 and 28 bp, with wild-type PCR products remaining uncut.
The relative proportion of wild type and mutant mtDNA genomes at each site was determined by the addition of 5 μCi [α-32P] dCTP (3000 Ci/mmol) prior to the last cycle of the PCR. Labelled products were digested with 10 U of the relevant restriction enzyme, separated through 12% non-denaturing polyacrylamide gels, and the radioactivity in each fragment quantified using ImageQuant software (Molecular Dynamics).
Analysis of cybrid clones
Total DNA was extracted from 55 cybrid clones by standard methods. These were screened for their level of m.7472Cins mutation by PCR-RFLP analysis, as described.
High-resolution northern blot analysis
The steady-state level of mt-tRNASer(UCN) was determined in patient III-7 muscle, and patient III-6 muscle, heart and brain, and in selected cybrid clones. The level of mt-tRNASer(UCN) was directly compared to the level of another mt-tRNA (mt-tRNALeu(UUR)).22 A PCR-generated mt-tRNASer(UCN) probe was amplified using the forward primer L7400 (positions 7400–7419) 5′-CCCACCCTACCACACATTCG-3′ and the reverse primer H7517 (positions 7517–7497) 5′-TTTGAAAAAGTCATGGAGGCC-3′. Following PhosphorImager analysis, the radioactive signal for the mt-tRNASer(UCN) probe was normalized to that of the mt-tRNALeu(UUR) probe for each sample.
Biochemical analysis of cybrid clones
Isolation of mitochondrial fractions from cultured clones and spectrophotometric assays of complexes I, II and IV, and citrate synthase activities were done as previously described.23
Galactose growth assay for assessing respiratory deficiency
Selected cybrid clones and corresponding parental and ρ° cell lines were plated in six-well plates at a density of 2.5 × 104 cells per well. Cells were grown in glucose-free DMEM supplemented with 10% FCS, 90 μg/ml galactose and 110 μg/ml sodium pyruvate. Cells were trypsinized from replicate plates after 3, 5, 7 and 10 days, total cell counts taken and averaged.
Polarographic analyses of intact cybrid clones
Oxygen consumption in intact cells was performed using a high-resolution oxygraph (Oroboros, Innsbruck). Cells were suspended in a Tris-based Mg2+-, Ca2+-deficient buffer (137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4 and 25 mM Tris-HCl, pH 7.4 at 25°C) and introduced into the oxygraph chamber. 2,4-Dinitrophenol was titrated until maximal uncoupling was achieved (30–60 μ M), and respiration was then inhibited by antimycin (20 nM). For substrate-specific respiration, a Tris-based permeabilization buffer was used (75 mM sucrose, 5 mM potassium phosphate, 40 mM KCl, 0.5 mM EDTA, 3 mM MgCl2, 0.35% (w/v) BSA and 30 mM Tris-HCl, pH7.4 at 25°C) and digitonin was added at a concentration of 10 μg/106 cells. Multiple substrate-inhibitor titrations with pyruvate (8 mM), malate (2 mM), glutamate (15 mM) and rotenone (100 nM), followed by succinate (10 mM) and antimycin (20 nM) were performed. Individual inhibitor-sensitive oxygen consumption rates were calculated as the time derivative of the oxygen concentration (DATLAB Analysis Software, Oroboros) and analysed using SPSS software. Data shown represent an average of a minimum of three determinations and are expressed in fmol O2/min per cell.
Mitochondrial protein synthesis in whole cultured cells
Mitochondrial protein synthesis was determined essentially as described previously,24 but with minor changes. Cells in exponential growth were plated into six-well plates and incubated overnight to achieve 60–75% confluency. Cells were washed with methionine/cysteine-free DMEM, and then incubated twice for 30 min at 37°C in 2 ml of the same medium. Following a 10 min incubation in methionine/cysteine-free DMEM supplemented with 10% dialysed FCS and 100 μg/ml emetine, ∼500 μCi 35S-cys methionine was added and incubation continued for a further 120 min before a further 10 min incubation in the presence of 15 μg cold methionine. The labelled cells were trypsinized, washed and resuspended in PBS containing 1 × protease inhibitors. Samples containing 50 μg of protein were electrophoresed through a 15% SDS-polyacrylamide gel, and the radioactivity in each sample detected using ImageQuant software (Molecular Dynamics).
Results
Histochemistry and biochemistry
Histochemical examination of the proband's muscle biopsy revealed a significant proportion (>80%) of cytochrome c oxidase (COX)-deficient fibres, many of these showing evidence of subsarcolemmal mitochondrial accumulation (ragged-red fibres) (Figure 2a). Similar observations were made in the muscle from her mother (patient II-5) and in several post-mortem tissues, including cardiac muscle, from her brother (patient III-7) (Figure 2b). Assessment of respiratory chain enzyme activities in the proband's skeletal muscle demonstrated marked deficiencies of both complex I and IV activities of 6 and 10% of normal controls, respectively. Complex II activity was normal (Table 1).

Sequential cytochrome c oxidase (COX)/SDH histochemistry of skeletal muscle from the proband (a) and the left ventricle of patient III-6 (b) showing a profound COX deficiency (blue stain) in many muscle fibres and evidence of mitochondrial proliferation. (c) Electropherogram highlighting the two sequence changes (m.7472Cins and m.7472A>C) present in the patient's muscle DNA, giving rise to a ‘C’ tract of eight nucleotides as opposed to six nucleotides in the wild-type sequence. (d) Quantification of the relative amounts of mutant and wild-type mtDNA in tissues from three family members for both the m.7472Cins mutation (upper panel) and the m.7472A>C transversion (lower panel). U, uncut sample; C, control; Cyb, 100% mutant m.7472Cins cybrid DNA; M, skeletal muscle; B, blood; Ur, urinary epithelial cells; Bu, buccal epithelial cells; F, fibroblasts.
mtDNA analysis
Direct sequencing of the entire mtDNA-coding region revealed several previously reported polymorphisms and two changes in the MTTS1 gene (Figure 2c). These were a previously reported pathogenic insertion of a single C nucleotide (m.7472Cins)9 and the recently reported A–C transversion (m.7472A>C),15, 16 which was not present in over 200 of our own control sequences or in two large databases of >2400 human mtDNA sequences.25, 26
PCR-RFLP analysis of the m.7472Cins mutation revealed widespread heteroplasmy in all tissues investigated from the patient and her clinically affected mother and brother, with highest levels in skeletal muscle (86, 84 and 83% mutant load, respectively) and lower levels in mitotic tissues such as blood, urine and fibroblasts (Figure 1). Assessment of other maternal relatives showed a clear segregation of m.7472Cins mutation load with clinical disease, while analysis of the m.7472A>C transversion revealed apparent homoplasmic levels of this mutation in every sample (Figures 1 and 2d). Single muscle fibre analysis was performed to determine whether the amount of the m.7472Cins mutation correlated with the observed biochemical phenotype, revealing substantially higher levels in COX-deficient fibres (93.2±0.80%, (n=8)) than in the COX-positive fibres (45.4±5.9, (n=10)), (P<0.0001, Student's t-test) (Figure 3a).

Confirmation of m.7472Cins pathogenicity (a) single fibre PCR-RFLP analysis of the m.7472Cins mutation in individual COX-positive and -deficient muscle fibres showing a clear segregation of high mutant load with the biochemical defect. (b) High-resolution northern blot of mt-tRNASer(UCN) steady-state levels in muscle from patient III-7 (1), and heart (2) and brain regions (3) from patient III-6, showing a virtual absence of the amount of the mature mt-tRNASer(UCN) transcript when compared to controls (labelled ‘C’).
mt-tRNA analysis in patient tissues
To investigate the processing of mt-tRNASer(UCN) from its precursor, the steady-state level of mt-tRNASer(UCN) was determined in the patient III-7's skeletal muscle, and in muscle, heart and brain of the patient III-6 by northern blot hybridization. Levels of the mature mt-tRNASer(UCN) transcript were undetectable when compared to controls, while the steady-state level of another mt-tRNA (mt-tRNALeu(UUR)) was unchanged (Figure 3b).
Isolation of transmitochondrial cybrid clones
A total of 55 cybrid clones were screened for m.7472Cins heteroplasmy by RFLP analysis. Three clones harbouring 100% m.7472Cins (DC164, DC26 and DC162), and three clones harbouring 0% m.7472Cins (DC167, DC159 and DC172) were chosen for further analysis. Alongside these, clones harbouring ∼25% (DC30), ∼50% (DC33) and ∼75% (DC113) m.7472Cins mutant loads were also investigated to determine whether any effect(s) observed segregated with the level of this mutation. All clones were shown to carry the m.7472A>C variant at homoplasmic levels.
Biochemical and genetic analysis of cybrid clones
We first investigated the growth of homoplasmic cybrid clones on galactose, a non-fermentable carbon source, to determine whether these expressed a respiratory deficiency. Cell counts were variable on days 3 and 5 between the two homoplasmic mutant clones assessed – DC167 (0% m.7472Cins, 100% m.7472A>C) and DC164 (100% m.7472Cins, 100% m.7472A>C) – but ultimately their fate was the same as the ρ°cells devoid of mtDNA, suggestive of a biochemical defect in both mutant cell lines (Figure 4a). Similar data were also obtained for clones DC159 and DC26 (not shown). Respiratory chain biochemical analysis of complexes with mtDNA-encoded subunits (complexes I, III and IV) showed a significant decrease in activity in clones with the m.7472A>C transversion alone (DC167 and DC172) when compared to that of the normal 143B parental control. However, in the clones harbouring both mutations at homoplasmic levels (DC164 and DC162), the decrease observed in biochemistry was much milder when compared to controls (Figure 4b). Results of respiration in intact cells revealed a similar pattern, where the clones with the m.7472A>C alone had a marked respiratory deficiency, whereas the clones harbouring both mutations respired at a rate comparable to controls (Figure 4c).

(a) Galactose growth assay confirmed the inability of the mutant clones to grow in glucose-free/galactose-supplemented medium over 10 days growth.143B ρ° cells are illustrated by the squares, 143B parental cells by triangles, clone DC167 by circles and clone DC164 by diamonds. Data shown are from a single experiment but were repeated (n=2) and the same findings replicated in different homoplasmic clones (DC159 and DC26) (b) Respiratory chain biochemistry (complex activities expressed as a ratio to the activity of citrate synthase activity, and expressed as a percentage of the activities of 143B parental cells (100%)) revealed a severe defect in those cells harbouring m.7472A>C alone; 143B parental cells (black bars), clone DC167 (grey bars), clone DC164 (white bars); data shown represent the mean of three determinations (±SEM) (c) Respiration in intact cells revealed a deficiency in O2 consumption in those cells harbouring m.7472A>C alone; 143B parental cells (black bars), clone DC167 (grey bars), clone DC164 (white bars). Data shown represent inhibitor-sensitive values as the mean of three determinations (±SEM) (d) In vitro translation of mitochondrial proteins in transmitochondrial cybrid clones cultured in emetine and pulse labelled for 2 h with 500 μCi 35S-cys methionine. Translation products were separated through a 15% SDS-polyacrylamide gel (see the Patient and methods) and are labelled on the basis of their molecular weight and previously reported data.24
Next, we investigated the steady-state levels of the mt-tRNASer(UCN) transcript in clones DC167, DC159 and DC172 (0% m.7472Cins, 100% m.7472A>C) and clones DC164, DC26 and DC162 (100% m.7472Cins, 100% m.7472A>C). Although the steady-state level of mt-tRNASer(UCN) was decreased in the clones harbouring only the m.7472A>C mutation, steady-state mt-tRNASer(UCN) levels were more severely decreased in the clones harbouring both mutations at homoplasmic levels (Figure 5a). Further to this result, we selected and grew other clones harbouring intermediate levels of the m.7472Cins mutation (∼25% (DC30), ∼50% (DC33) and ∼75% (DC113)) in order to assess whether the steady-state mt-tRNASer(UCN) levels changed with increasing mutation load. As the level of m.7472Cins increases, the steady-state level of mt-tRNASer(UCN) decreases with cells harbouring >50% m.7472Cins mutation exhibiting barely detectable transcript levels (Figure 5b). These data would suggest that both the m.7472Cins and m.7472A>C mutations act synergistically in vitro, with the suppressive effect of the m.7472A>C mutation regulated at the level of transcription by the m.7472Cins mutation load.

(a) High-resolution northern blot showing the steady-state level of mt-tRNASer(UCN) is reduced in transmitochondrial cybrid clones harbouring 100% m.7472A>C, and is severely reduced in clones harbouring both m.7472A>C and m.7472Cins at homoplasmic levels (b) High-resolution northern blot showing the level of mt-tRNASer(UCN) clearly decreases as the level of m.7472Cins increases; all clones are homoplasmic for m.7472A>C. Levels of mt-tRNASer(UCN) compared to controls are shown (upper panel). PCR-RFLP analysis confirmed the level of m.7472Cins heteroplasmy in the clones analysed (lower panel).
Mitochondrial protein synthesis
Whole cell 35S-labelling experiments showed that mitochondrial protein synthesis in clone DC164 (100% m.7472Cins+100% m.7472A>C) was comparable to that of parental cells, while clone DC167 (0% m.7472Cins+100% m.7472A>C) exhibited a marked reduction in the synthesis of all mitochondrial proteins (Figure 4d). These results were shown to be reproducible both in these and other (DC159 and DC26) clones (not shown).
Discussion
The MTTS1 gene is a hot spot for pathogenic mutations exhibiting phenotypic differences. Many mutations, including m.7472Cins, are associated with a phenotype consisting predominantly or exclusively of deafness. Neurological features including myoclonus, ataxia and monomelic amyotrophy have been reported in families with the m.7472Cins mutation at high levels of heteroplasmy, yet the histochemical and biochemical defect may be subtle.9, 10, 27
We report a family with mitochondrial disease and clear evidence of maternal transmission of their clinical abnormalities and mtDNA mutation. All individuals show phenotypic variation both from previously reported cases with the m.7472Cins mutation alone, and those with both the m.7472Cins and the m.7472A>C mutations together. The factors determining this are largely unresolved, but the segregation of mutation load, nuclear genetic factors and mtDNA sequence variation may all contribute as modifiers of phenotype. As examples, previous studies have reported a transmitochondrial cybrid line with high levels of the m.3243A>G MTTL1 mutation and an additional heteroplasmic, novel anticodon mutation in the MTTL2 gene, predicted to generate a suppressor mt-tRNA capable of decoding UUR leucine,28 while the expression of the m.7445A>G deafness mutation can be influenced by distant mtDNA polymorphisms that affect the folding of the primary mtDNA transcript and consequently processing.13 It is not unreasonable to speculate therefore that the m.7472A>C mutation, which was detected at homoplasmic levels in all family members, is acting to modify the pathogenicity of the m.7472Cins mutation, thereby explaining the dramatic loss of steady-state mt-tRNASer(UCN) transcript and marked COX deficiency in patient tissue that is not apparent in patients harbouring the m.7472Cins mutation alone. While the reduction of steady-state levels of mt-tRNASer(UCN) in homoplasmic m.7472Cins transmitochondrial cybrids is thought to be due to altered processing,12, 14 it is possible that the m.7472A>C is acting in synergy to alter mt-tRNA stability. Our data from cybrids homoplasmic for the m.7472A>C mutation would certainly support the notion that this mutation alone is sufficient to cause a biochemical defect. Both previous reports documenting the m.7472Cins mutation in association with the m.7472A>C have proposed a role for the m.7472A>C mutation in modifying the phenotype of m.7472Cins,15, 16 although both suggest that the mutation is polymorphic. Homoplasmy, as seen in our family is indeed a common feature of mtDNA polymorphic variants, but we provide sufficient data here to argue that this sequence variation is not a neutral polymorphism.
The m.7472A>C mutation has never been reported without association with the m.7472Cins mutation – its absence from control databases argue against it being a common polymorphic variant. By isolating transmitochondrial cybrid clones with the m.7472A>C transversion in both the presence and absence of the m.7472Cins mutation, we have attempted to clarify the precise role of this homoplasmic variant. All clones are in the 143B osteosarcoma cell line to remove any influence of differences in nuclear background. Clones were selected for their appropriate levels of homoplasmy/heteroplasmy, therefore controlling for any influence of heteroplasmy. Northern blot analysis clearly revealed that the steady-state level of mt-tRNASer(UCN) is reduced in both clones when compared to controls, but is far more severely reduced in the clones with both mutations than in clones with just the m.7472A>C mutation, a pattern consistent with that observed in vivo when mt-tRNASer(UCN) levels are compared in patients with the m.7472Cins mutation to those with both mutations. Results of growth assays on cybrids are consistent with this observed reduction in mt-tRNASer(UCN), in which we confirm clones with either m.7472A>C alone, or in association with m.7472Cins show a defect when grown under conditions of respiratory stress. However, surprisingly results from respiratory chain biochemistry, respiration and protein synthesis analysis show a severe defect when compared to normal controls only in cell lines harbouring the m.7472A>C mutation alone, an unexpected result when compared to high-resolution northern blot and growth assay analysis of the same clones.
Although the pattern seen in vitro in the steady-state level of mt-tRNASer(UCN) remains consistent with what we see in vivo, the reduction in levels appears more severe in vivo. Our clones harbouring both mutations have a steady-state level of mt-tRNASer(UCN) of ∼5% of the control cell line, and respiratory chain biochemistry and respiration is mild to comparable, respectively, to that of controls. This implies that remaining mt-tRNASer(UCN) transcript in these cells is sufficient to maintain respiratory function at a normal level. We see a much more severe reduction in mt-tRNASer(UCN) and subsequent deficiency in biochemistry in vivo. Indeed, transmitochondrial cybrids homoplasmic for m.7472Cins have ∼40% steady-state levels of mt-tRNASer(UCN) and only a mild abnormality in mitochondrial protein synthesis.11 In this regard, the virtual absence of the mt-tRNASer(UCN) transcript in the proband's muscle, coupled with the severe histochemical and combined biochemical deficiencies of complexes I and IV are unprecedented in families previously described with this mutation, though entirely compatible with the more devastating clinical presentation in this family. However, we must appreciate the differences observed between in vitro to those seen in vivo, and be cautious in extrapolating this data as being entirely conclusive. Mitochondrial respiratory chain function is dependent on the co-ordination of both the nuclear and mitochondrial genomes, and as such different transmitochondrial cybrid nuclear backgrounds may give different results. It is well recognized that the immortalized tumour-derived 143B osteosarcoma cells are aneuploid and can exhibit marked differences in chromosomal make-up, even in clonal cultures, and this may in part explain the difference in results observed between different laboratories working on cybrids with the same nuclear background.
Perhaps the most convincing data arguing the m.7472A>C is not polymorphic, is the marked reduction in mt-tRNASer(UCN) transcript, and the severe deleterious effects on biochemistry, respiration and mitochondrial protein synthesis in vitro that occurs in the presence of the m.7472A>C mutation alone. Single fibre data appeared to confirm the primary pathogenic mutation to be the m.7472Cins. As the m.7472A>C was homoplasmic in all patient tissues analysed, this mutation clearly does not segregate with observed phenotype, but arguably has an effect when studied in vitro. A significant limitation is that as this mutation has never been observed alone in a patient, no comparison can be made between our in vitro result and what would occur in vivo. From this data, it would not be unreasonable to suggest that it is in fact the seventh ‘C’ residue at this position in the mt-tRNASer(UCN) molecule, and not the extra base length created by m.7472Cins that is important for pathogenesis. The further addition of a ‘C’ residue in the tract created by m.7472A>C, resulting in eight consecutive ‘C’ residues appears to be less detrimental than seven adjacent ‘C’ bases at this position.
It is interesting to note that both previous reports of the two mutations together have observed milder effects in their patients to those harbouring the m.7472Cins alone.15, 16 We observed a more severe effect, for example severe COX deficiency in muscle biopsies, and myopathy leading to death by cardiac failure, and would argue that the findings in the previous reports of the m.7472A>C are not so dissimilar. Like our case, Pulkes et al15 describe a family with myopathic presentation with no CNS involvement, and a high (>90%) COX deficiency, with a high proportion of ragged-red fibres. This is a more severe histochemical finding than in their patient with just the m.7472Cins. The patient described in Cardaioli et al died at a young age (32) from rapid progressive encephalomyopathy, a similar clinical picture to other cases with just the m.7472Cins,10 and like our case resulting in death. This patient had 75% COX deficiency on histochemical analysis. Only mild COX deficiency is usually seen in cases of m.7472Cins,12, 27 and RRF are rarely seen;27, 29 to date all patients described with the m.7472A>C mutation have a high proportion of COX-deficient fibres and RRF. In this respect, it could be argued that the presence of the m.7472A>C decreases the ‘pathogenic effect’ of m.7472Cins, in that higher levels can be tolerated in muscle. The differences between the reported families remain unresolved and can only be assumed to be due to other factors, for example environmental differences, differences in nuclear background or possibly other variations in their mtDNA.
In summary, the presence of the m.7472A>C clearly influences the expression of the m.7472Cins when studied in vitro. Although it seems to suppress the effects of the mutation in functional assays (eg biochemistry and respiration), it clearly enhances the effects of the mutation in terms of reducing the steady-state levels of the affected mt-tRNA. This highlights just how complex the mechanisms of pathogenesis are in mtDNA mutations, although it remains unclear if the differences found in these cells are truly representative of what is happening in vivo. The experiments in cybrid cells have been repeated on several occasions in numerous clones, and while we are confident in the ability of our assays to detect changes in mitochondrial respiration, biochemical function and mt-tRNA levels, it does also emphasize the inherent difficulties of some cellular models, and relating the changes observed in culture to the findings in the patient. Our study not only highlights the clinical and genetic diversity of mitochondrial disease and difficulties of assigning pathogenicity to specific mt-tRNA variants, but also that we should not underestimate the role that other sequence changes in the mitochondrial genome, albeit homoplasmic, might play in the expression of pathogenic mt-tRNA mutations.
References
DiMauro S, Schon EA : Mitochondrial DNA mutations in human disease. Am J Med Genet 2001; 106: 18–26.
Taylor RW, Turnbull DM : Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005; 6: 389–402.
Brown MD, Starikovskaya E, Derbeneva O et al: The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup J. Hum Genet 2002; 110: 130–138.
Qu J, Li R, Zhou X et al: The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci 2006; 47: 475–483.
Reid FM, Vernham GA, Jacobs HT : A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat 1994; 3: 243–247.
Hutchin TP, Parker MJ, Young ID et al: A novel mutation in the mitochondrial tRNASer(UCN) gene in a family with non-syndromic sensorineural hearing impairment. J Med Genet 2000; 37: 692–694.
Sue CM, Tanji K, Hadjigeorgiou G et al: Maternally inherited hearing loss in a large kindred with a novel T7511C mutation in the mitochondrial DNA tRNASer(UCN) gene. Neurology 1999; 52: 1905–1908.
Verhoeven K, Ensink RJ, Tiranti V et al: Hearing impairment and neurological dysfunction associated with a mutation in the mitochondrial tRNASer(UCN) gene. Eur J Hum Genet 1999; 7: 45–51.
Tiranti V, Chariot P, Carella F et al: Maternally inherited hearing loss, ataxia and myoclonus associated with a novel point mutation in mitochondrial tRNASer(UCN) gene. Hum Mol Genet 1995; 4: 1421–1427.
Schuelke M, Bakker M, Stoltenburg G, Sperner J, von Moers A : Epilepsia partialis continua associated with a homoplasmic mitochondrial tRNASer(UCN) mutation. Ann Neurol 1998; 44: 700–704.
Toompuu M, Tiranti V, Zeviani M, Jacobs HT : Molecular phenotype of the np 7472 deafness-associated mitochondrial mutation in osteosarcoma cell cybrids. Hum Mol Genet 1999; 8: 2275–2283.
Toompuu M, Yasukawa T, Suzuki T et al: The 7472insC mitochondrial DNA mutation impairs the synthesis and extent of aminoacylation of tRNASer(UCN) but not its structure or rate of turnover. J Biol Chem 2002; 277: 22240–22250.
Guan MX, Enriquez JA, Fischel-Ghodsian N et al: The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol Cell Biol 1998; 18: 5868–5879.
Toompuu M, Levinger LL, Nadal A, Gomez J, Jacobs HT : The 7472insC mtDNA mutation impairs 5′ and 3′ processing of tRNASer(UCN). Biochem Biophys Res Commun 2004; 322: 803–813.
Pulkes T, Liolitsa D, Eunson LH et al: New phenotypic diversity associated with the mitochondrial tRNASer(UCN) gene mutation. Neuromuscul Disord 2005; 15: 364–371.
Cardaioli E, Da Pozzo P, Cerase A et al: Rapidly progressive neurodegeneration in a case with the 7472insC mutation and the A7472C polymorphism in the mtDNA tRNAser(UCN) gene. Neuromuscul Disord 2006; 16: 26–31.
Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG : The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain 2003; 126: 413–423.
Old SL, Johnson MA : Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle. Histochem J 1989; 21: 545–555.
Kirby DM, Thorburn DR, Turnbull DM, Taylor RW : Biochemical assays of respiratory chain complex activity. Methods Cell Biol 2007; 80: 93–119.
King MP, Attardi G : Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989; 246: 500–503.
Taylor RW, Taylor GA, Durham SE, Turnbull DM : The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res 2001; 29: E74.
Taylor RW, Giordano C, Davidson MM et al: A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol 2003; 41: 1786–1796.
Pye D, Kyriakouli DS, Taylor GA et al: Production of transmitochondrial cybrids containing naturally occurring pathogenic mtDNA variants. Nucleic Acids Res 2006; 34: e95.
Chomyn A : In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol 1996; 264: 197–211.
Brandon MC, Lott MT, Nguyen KC et al: MITOMAP: a human mitochondrial genome database – 2004 update. Nucleic Acids Res 2005; 33: D611–D613.
Ingman M, Gyllensten U : mtDB: human mitochondrial genome database, a resource for population genetics and medical sciences. Nucleic Acids Res 2006; 34: D749–D751.
Fetoni V, Briem E, Carrara F, Mora M, Zeviani M : Monomelic amyotrophy associated with the 7472insC mutation in the mtDNA tRNASer(UCN) gene. Neuromuscul Disord 2004; 14: 723–726.
El Meziane A, Lehtinen SK, Hance N et al: A tRNA suppressor mutation in human mitochondria. Nat Genet 1998; 18: 350–353.
Jaksch M, Klopstock T, Kurlemann G et al: Progressive myoclonus epilepsy and mitochondrial myopathy associated with mutations in the tRNASer(UCN) gene. Ann Neurol 1998; 44: 635–640.
Acknowledgements
We thank Christine Hayes, Karen Ayyad, Nadine Romain and Winsome Walker for excellent technical assistance. This work was supported by grants from the Muscular Dystrophy Association and the United Mitochondrial Disease Foundation (to TT and RGH), the US National Institutes of Health (to MMD) and The Wellcome Trust, Muscular Dystrophy Campaign and Newcastle upon Tyne Hospitals NHS Foundation Trust (to DMT and RWT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Swalwell, H., Blakely, E., Sutton, R. et al. A homoplasmic mtDNA variant can influence the phenotype of the pathogenic m.7472Cins MTTS1 mutation: are two mutations better than one?. Eur J Hum Genet 16, 1265–1274 (2008). https://doi.org/10.1038/ejhg.2008.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.65
Keywords
This article is cited by
-
Two mitochondrial DNA polymorphisms modulate cardiolipin binding and lead to synthetic lethality
Nature Communications (2024)
-
Mitochondrial Disease in Childhood: mtDNA Encoded
Neurotherapeutics (2013)
