
Abstract
Pendred syndrome (PS) and DFNB4, a non-syndromic sensorineural hearing loss with enlargement of the vestibular aqueduct (EVA), are caused by mutations in the SLC26A4 gene. Both disorders are recessive, and yet only one mutated SLC26A4 allele, or no mutations, are identified in many cases. Here we present the genetic characterization of 105 Spanish patients from 47 families with PS or non-syndromic EVA and 20 families with recessive non-syndromic hearing loss, which segregated with the DFNB4 locus. In this cohort, two causative SLC26A4 mutations could be characterized in 18 families (27%), whereas a single mutated allele was found in a patient with unilateral hearing loss and EVA in the same ear. In all, 24 different causative mutations were identified, including eight novel mutations. The novel p.Q514K variant was the most prevalent mutation in SLC26A4, accounting for 17% (6/36) of the mutated alleles identified in this study, deriving from a founder effect. We also characterized a novel multiexon 14 kb deletion spanning from intron 3 to intron 6 (g.8091T_22145Cdel). This study also revealed the first case of a de novo recessive mutation p.Q413P causing PS that arose in the proband's paternal allele, the maternal one carrying the p.L445W. The relevance of our results for genetic diagnosis of PS and non-syndromic EVA hearing loss is discussed.
Similar content being viewed by others


Introduction
Mutations in the SLC26A4 gene cause Pendred's syndrome1 (PS; MIM#274600) and DFNB4,2 the latter being a non-syndromic form of hearing loss (MIM#600791) presenting with enlargement of the vestibular aqueduct (EVA). Pendred syndrome (PS) is an autosomal recessive condition characterized by sensorineural deafness and goitre which may account for up to 10% of profound childhood hearing impairment.3 Patients with PS typically suffer from pre- or peri-lingual hearing loss and they have congenital inner ear malformations ranging from EVA to Mondini malformation. Goitre is not a constant feature in PS and it only occurs in about one-third of patients who typically have abnormal iodide release by the thyroid on the perchlorate discharge test. In contrast, other patients with sensorineural hearing loss and EVA do not develop thyroid disease (non-syndromic EVA).
The SLC26A4 gene (NM_000441) encodes pendrin (780 amino acids), a member of the SLC26 family of anion transporters.1 Functional analysis showed pendrin is an apical transporter of iodide in thyroid cells,4, 5 and it is likely to mediate secretion of HCO3– into the endolymph in the cochlea.6 Pendrin also mediates chloride/base exchange in the kidney cortex.7 Following the identification of the SLC26A4 gene, the mutations in SLC26A4 causing PS and non-syndromic EVA began to be characterized and more than one hundred mutant alleles have now been identified, mostly missense mutations. Some mutations are recurrent and have been identified in several populations, but many others have only been described in one family.8, 9, 10, 11, 12, 13, 14, 15 Moreover, in many patients with PS or non-syndromic EVA only one mutation has been identified in the SLC26A4 coding region while in others, no mutations at all have been identified in the coding sequence. Previous studies provide evidence that PS and DFNB4 hearing loss are phenotypic variants of the same recessive genetic disorder, and other studies indicate non-syndromic EVA is a heterogeneous genetic condition which is often caused by a single mutation in SLC26A4 in combination with other genetic or environmental factors.10 Furthermore, non-syndromic EVA was recently shown to segregate with digenic inheritance, in a patient carrying a mutation in the SLC26A4 gene and another in FOXI1 (MIM#601093), a gene encoding a transcriptional activator of SLC26A4.16
To gain a better understanding of the genetic basis of PS and non-syndromic EVA, we analysed 67 unrelated Spanish hearing-impaired families. The spectrum of SLC26A4 mutations in our population involved mostly missense mutations and one multiexon deletion. We report on the prevalence of SLC26A4 mutations in our cohort as well as the genotype–phenotype associations.
Subjects and methods
Subjects
A total of 105 patients from 67 unrelated families were included in this study that were divided into three groups on the basis of the clinical, audiological and genetic findings as follows: (A) 21 PS patients from 12 different families, all patients in this group display inner ear malformations including EVA or EVA with hypoplasia of the cochlea (Mondini malformation), goitre, and/or a positive result in the perchlorate test; (B) 43 cases with non-syndromic EVA from 35 unrelated families. In this group, 35 patients had EVA and eight had Mondini malformation, none developed goitre and the perchlorate test was normal in the nine cases on whom it was performed; (C) 41 patients from 20 unrelated families with non-syndromic hearing impairment segregating with the DFNB4 locus, with no other available clinical data. These cases were selected from 115 families after genotype analysis with STR markers linked to the DFNB4 locus.
Mutations in the DFNB1 locus, the p.Q829X mutation in the OTOF gene and the mitochondrial 1555A>G mutation were ruled out as the genetic cause of deafness in all patients. Blood samples were obtained from all individuals after obtaining their written informed consent. DNA was purified from these by standard procedures.
Clinical examinations
Spanish patients with hearing impairment were referred to our laboratory for molecular diagnosis, having already undergone physical and age-appropriate audiological examinations. Additional clinical examinations included computed tomography of the temporal bones, ultrasonography to detect goitres, functional thyroid tests including serum-free thyroxine and thyrotropin, and the perchlorate discharge test to reach a firm diagnosis of PS. The perchlorate discharge test involved the oral administration of 131Iodide and the uptake of radioiodide in the thyroid was measured 2 h later, before potassium perchlorate was administered orally. The perchlorate discharge test was considered positive when 1 h after administration of the perchlorate, the amount of radioiodide in the thyroid decreased by more than 10% relative to the initial uptake.
Genotyping
Haplotype analysis was performed with STR markers, linked to the DFNB4 locus on 7q31 (Cen-D7S501-D7S2420-D7S496-D7S2459-D7S3312-D7S3074-D7S692-D7S2425-Tel). D7S2459 and D7S3312 are intragenic microsatellites located in intron 10. To exclude alternative paternity in patient E343.1 who carries the de novo p.Q413P mutation, the members of the family were genotyped with markers linked to the DFNB4 locus described above and with 17 STR markers located on chromosomes 2, 3, 10, 13 and 22 (data not shown).
SLC26A4 mutation screening
PCR amplification of the 20 exons coding for pendrin (2–21) and their flanking intronic sequences was performed with primers designed with Oligo v6.67 software (primer sequences and PCR conditions are available on request). PCR products were screened for mutations by DHPLC (Denaturing High Performance Liquid Chromatography) on a Wave™ DNA Fragment Analysis System (Transgenomic™, USA). All variant profiles were characterized by sequencing the product of a second PCR amplification using an ABI PRISM 3100 Genetic Analyser (Applied Biosystems). The two FOXI1-binding transcriptional regulatory elements in SLC26A4 exon 1 were sequenced directly in each PS patient and in the ‘monoallelic’ E595.1 patient. The parental origin of the mutations was ascertained by direct sequencing of the variant PCR product from the parents' DNA. Each SLC26A4 sequence variant was tested in 214 unrelated normal-hearing controls by DHPLC, following the same procedure. Sequence variants were also tested in 144–341 Spanish patients with non-syndromic hearing impairment (NSHI). Mutation numbering is based on the SLC26A4 cDNA sequence from Genbank (NM_000441), with the A of the translation start codon considered as nucleotide number 1.
Cloning of the de novo p.Q413P mutation
A SLC26A4 genomic fragment spanning from exon 10 to exon 11 (4485 pb) was amplified by long-range PCR from the genomic DNA of patient E343.1. The PCR product was purified and cloned into the pSMART-cDNA vector (Lucigen Corporation). Subsequently, several recombinant plasmids were isolated and characterized by genotyping the intronic markers D7S3312 and D7S2459, and sequencing exons 10 and 11.
Characterization of the SLC26A4 multiexon deletion
Haplotype analysis of family S129 was performed on genomic DNA by PCR amplification of 17 SLC26A4 single nucleotide polymorphisms located in introns 2–6, 8, 12, 14, 17–21 and taken from the NCBI SNP database. The amplified PCR products were then sequenced directly.
SLC26A4 gene dosage analysis of exons 1–10 was performed by real-time quantitative PCR using the Power SYBR® Green PCR Master Mix (Applied Biosystems). Absolute quantification of amplicons was established by comparing the lowest threshold cycle (CT) of the real-time PCR samples against a standard curve of known copy numbers. To map the deletion breakpoints, long range PCR was performed on genomic DNA using the Expand Long Template PCR System, with primers located at intron 2 and intron 7. A 6 kb PCR product was amplified with DNA from patient S129.2, which was later characterized by restriction mapping with the enzymes Bam HI, Bgl II and Cla I. Since there was a single Bgl II site in the PCR product, we performed a novel PCR using an upper primer located up-stream of the Bgl II site and a lower primer at intron 7. The PCR product of approximately 2 kb was directly sequenced to characterize the breakpoint junction of the deletion. We also designed a test to detect the multiexon deletion by PCR amplification using primers DEL14 Kb-U (5′-AACAACTCCGAGAAGACC-3′) and DEL14 Kb-L (5′-AAGATGTCTTAAAAGAAAAAATG-3′), which produce a 314 bp fragment from the mutated allele and no PCR product from the wild type (Figure 1).

(a) Genomic organization of the SLC26A4 gene around the deletion breakpoints. Exons, repetitive sequence elements and primers used for PCR amplification of the 6 and 2 kb fragments are indicated. The sequence electropherogram of the deletion junction fragment is shown in the upper panel. (b) PCR assay for testing the novel deletion. The presence of the deleted allele is reflected by a PCR product of 314 bp derived from the mother and the siblings of family S129. (c) MAR potential of SLC26A4 regions around the deletion breakpoints according to the Mar-Finder algorithm. Coordinate numbering is based on the genomic sequence of SLC26A4 (NT_007933).
In silico analysis
The evolutionary conservation of pendrin residues among ten SLC26A4 orthologues was investigated at the ClustalW EMBL-EBI web site (http://www.ebi.ac.uk/clustalw/). The pathogenicity of the intronic mutations was investigated using the BDGP (Berkeley Drosophila Genome Project) Splice Site Prediction program (http://www.fruitfly.org/seq_tools/splice.html). RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/) was used to search for repetitive elements in introns 3 and 6. MAR-Wiz (http://www.futuresoft.org/) was used to search for scaffold/matrix attachment regions (S/MARs). Palindromic elements were searched for with Palindrome (http://bioweb.pasteur.fr/seqanal/interfaces/palindrome.htlm) and PatSearch (http://www.ba.itb.cnr.it/BIG/PatSearch/) was used to search for DNA sequence motifs known to be associated with deletion and translocation breakpoint junctions.17
Results
SLC26A4 mutation screening
Our cohort of 105 Spanish patients from 67 unrelated families included 12 families with PS, 35 with non-syndromic EVA and 20 families with hearing impairment that segregated with the DFNB4 locus. In this cohort we identified a total of 26 different sequence variants (Table 1), 9 of which were novel. Most of the sequence variants are missense changes (18/26, 69.2%), six of which have not been described previously: p.V88I; p.P140H; p.Q413P; p.G424D; p.T485R; and p.Q514K. We also identified three frameshift mutations, one of which is the novel c.1238delA mutation that changes the Gln413 to Arg creating a premature stop codon after 54 amino acids. Furthermore, four splice site variants were detected, one of which is novel (c.1001+5G>T). The splice site prediction program used indicated that c.1001+5G>T would significantly affect splicing decreasing the score of the exon 8 splice donor site from 0.98 in the wild type to 0.21 in the mutated variant. None of the novel changes were identified in 214 normal-hearing Spanish controls with the exception of p.V88I (1/214). Furthermore, the novel missense changes p.P140H, p.Q413P, p.G424D, p.T485R and p.Q514K affected residues conserved among all 10 orthologues and thus we assumed that they are pathogenic mutations. By contrast, we considered p.V88I to be a low frequency polymorphism, since Val88 is not conserved and Ile occurs at position 88 in several SLC26A4 orthologues. Indeed this change arose in cis in the maternal chromosome of patient S226.1 along with the p.R409H mutation.14 The previously reported p.G740V mutation in patient S707.1 was identified in cis with the p.T307M mutation, as found in a French patient.13 Gly at residue 740 is not conserved in SLC26A4 orthologues and we found the previously reported p.G740S variant (NCBI SNP database, rs17154353) in one normal-hearing Spanish control. Hence, we concluded that the allelic variants p.G740V and p.G740S are rare polymorphisms.
SLC26A4 mutated alleles were tested in the group of unrelated Spanish patients with NSHI. This analysis detected a patient with the p.Q514K mutation, one with the p.D724G variant, and another carrying the p.G740V polymorphism (Table 1). In all, 39 independent alleles were identified in this study, 36 of which are pathogenic.
The p.Q514K mutation was identified in six independent alleles (Table 1 and 2) and it was the most prevalent mutation, accounting for 17% (6/36) of the pathogenic alleles identified in this study. All six Spanish p.Q514K chromosomes shared a core haplotype defined by alleles 132, 146, 190 and 182 of markers D7S496, D7S2459, D7S3312, and D7S3074, respectively (Table 2). When assessed using a Fisher's exact analysis, the p.Q514K mutation was associated with the core haplotype (P<0.001), suggesting that the p.Q514K chromosomes derived from a common founder.
Our mutational analysis of SLC26A4 carried out does not detect intragenic deletions of the gene that may occur in the heterozygous state, so we further investigated the presence of exon deletions in family S129, with two PS siblings, in whom only a paternal p.T485R mutation was found. Members of the family were genotyped for a set of 17 single nucleotide polymorphisms along the SLC26A4 gene and the segregation analysis revealed a loss of heterozygosity for the rs2712212 SNP in intron 6, for which the siblings did not inherit the maternal allele. Indeed, a quantitative analysis of SLC26A4 gene dosage, carried out by real-time PCR, confirmed that there was a reduced dose of exons 4 to 6 in the DNA from the mother and her two children. To determine the extent of the deletion, a long PCR assay was performed on the mother's DNA to amplify the SLC26A4 gene from intron 2 to intron 7. The fragment identified in this assay was only 6 kb, smaller than the 20 kb fragment expected from the wild type allele. Restriction analysis of the PCR product defined the intervals where the breakpoints of the deletion might be located. Thus, we developed a robust PCR assay that produces a fragment of 2 kb and direct sequencing of this fragment revealed the breakpoint junction (Figure 1). Hence, we mapped the 5′ and 3′ deletion breakpoints at nucleotide positions g.8091 (intron 3) and g.22145 (intron 6) respectively. The novel g.8091T_22145Cdel removes a total of 14052 bp from the SLC26A4 gene and disrupts the open reading frame, which would result in the truncation of the protein at residue 105. We developed a specific PCR assay to test this multiexon deletion (Figure 1b) and after deletion screening the cohort of 67 patients, as well as 144 unrelated Spanish patients with NSHI, no other case carrying the novel deletion was found.
Sequence analysis of the regions surrounding the breakpoints demonstrated an array of repetitive elements with different similarities to the respective consensus sequences (Figure 1a). Also, seven topoisomerase I consensus cleavage sites (CTT, GTT, AAT and GAT) were detected, two of which (GTT and AAT) were located at the 5′ and 3′ breakpoints, respectively. However, no extensive sequence homology between the breakpoint regions was found, ruling out unequal homologous recombination as the mechanism responsible for the deletion. Further analysis of the sequence over the entire deleted region indicated that the 3′ breakpoint is flanked by an S/MAR (Scaffold attachment/matrix-associated region), with a high MAR potential (Figure 1c).
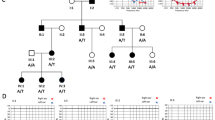
In the PS patient E343.1, we identified two pathogenic mutations: p.L445W (c.1334T>G in exon 11) and p.Q413P (c.1238A>C in exon 10). However, the haplotype analysis of members of the family did not coincide with a segregation of deafness with the SLC26A4 locus since the patient and his unaffected sister inherited the same parental haplotypes (Figure 2a). Mutational analysis showed that patient E343.1 inherited p.L445W from his mother; however, p.Q413P was not found in DNA samples from either parent or from the unaffected sister, indicating that p.Q413P was a de novo mutation. After excluding alternative paternity, we investigated the parental origin of the mutation in exon 10 by performing long-range PCR from exon 10–11 on genomic DNA from the patient. Genetic analysis of recombinant clones showed de novo mutation p.Q413P (c.1238A>C) occurred in the paternal allele, (Figure 2b). Indeed, the two missense mutations identified in the patient occurred in different parental alleles, in accordance with the recessive pattern of inheritance of PS/DFNB4 hearing loss.

(a) Haplotypes of family E343. Segregation analysis was performed with 8 STR markers linked to the SLC26A4 locus used to haplotype the families. Numbers indicate the sizes of the alleles in base pairs. (b) Genetic analysis of recombinant clones derived from the patient. Bars represent the cloned SLC26A4 genomic fragments spanning from exon 10 to exon 11. Allele sizes of the intragenic markers D7S2459 and D7S3312 in base pairs (upper panel) are indicated and sequence electropherograms showing the de novo c.1238A>C mutation in the allele derived from the father and the c.1334T>G in the maternal allele.
Genotype and phenotype correlation
We screened 21 patients from 12 independent families with a clinical diagnosis of PS for mutations in SLC26A4. Within this group, we detected the two parental SLC26A4 mutations in eight families (60%), while in the remaining four cases no mutation was found (Table 3). It is noteworthy that in the patients from three of the families that carried no mutations (S353, E67, and E412) the values of the discharge in the perchlorate test (12–16%) were close to the normal threshold (10%), while none of them developed goitre or displayed abnormal thyroid function (T4 and thyrotropin test). The fifth patient (S665.1) in whom no mutations were detected, at the age of 7, had a mild unilateral deafness in the right ear and unilateral Mondini malformation on the same side with positive perchlorate test (47%). Unilateral clinical presentation is rare in PS and since no SLC26A4 mutations were found in patient S665.1, the initial diagnosis of PS may not be accurate.10
On the other hand, six (17%) of the 35 independent cases with non-syndromic EVA were found to carry two SLC26A4 mutations while only one mutation was identified in patient E595.1, a sporadic case where unilateral cophosis was observed with EVA. Once again, a unilateral clinical defect was not associated with two mutations. Moreover, two mutated SLC26A4 alleles were detected in four (20%) of the 20 familial cases (S9, S461, S471 and S707) in which sensorineural hearing loss segregated with the DFNB4 locus. Clinical revision of several of these patients showed phenotypic traits of PS and non-syndromic EVA (Table 3). Taking all together, in our group of 67 unrelated families, 18 (27%) had two pathogenic mutations and one (1.5%) case carried only one mutated allele. Furthermore, we identified two patients carrying only one mutated allele p.Q514K, p.D724G in the group of patients with NSHI (Table 1). Since none of these patients had inner ear malformations, it is likely that they are coincidental carriers of the SLC26A4 mutations and that their deafness is caused by mutations at other loci.
No genotype–phenotype correlation could be attributed to different SLC26A4 mutations. However, if we compare the effect of two different classes of mutations, truncating mutations versus missense mutations, we found two patients from the S283 family who carried the c.279delT in homozygous state and had the most severe phenotype with respect to hearing loss (Table 3).
Discussion
Mutational analysis of the SLC26A4 gene on our cohort of 67 unrelated Spanish families identified 26 different variants (18 missense changes, three frameshift mutations, four splice site variants and one multiexon deletion), nine of which are novel. The pathogenic effect of the missense variants was assessed on the basis of two main criteria: the evolutionary conservation of the affected residue and its occurrence in a large control cohort of 214 normal hearing Spanish subjects. Thus, we concluded that p.V88I and p.G740V are rare, non-pathogenic polymorphisms. Two previously described missense mutations, p.E29Q and p.D724G, were found each one in a Spanish patient with non-syndromic EVA who carried another mutated allele in trans. However, one p.E29Q allele and two p.D724G alleles were also found in the 214 normal-hearing control population. Both mutated residues are conserved in all the mammalian orthologues, although the Glu at position 29 is not conserved in Danio rerio. The p.E29Q allele has been reported in two French patients9, 13 and in other three cases of unknown ethnic origin11, 20, 24 and except in one patient, it is accompanied by a second mutation. The p.D724G mutation has only been described in one patient of unknown ethnic origin who also carried a second mutation,20 as well as in our previously published Spanish family.23 We did not find the p.D724G variant in the first screening of 70 healthy controls, but it was found twice when we augmented the size of the control population to 214 individuals. Although it is possible that the normal-hearing controls carrying these mutations may be heterozygous carriers, it seems advisable to further assess the deleterious effect of these missense changes.
Here we define the breakpoints of an intragenic deletion in the SLC26A4 gene spanning from intron 3 to intron 6. Other two intragenic deletions (IVS2-IVS3del4 kb8 and IVS4-IVS6del25) have been previously described, but the deletion breakpoints were not characterized. Thus, the 14052 bp deletion found in the Spanish patient is a novel occurrence. Characterizing the deletion breakpoints allowed us to design a simple PCR assay to test for the novel multiexon deletion. This test did not detect any other case carrying this deletion in our group of Spanish patients. However, it would be advisable to use this test to screen other populations, especially the patients in whom only one mutated allele has been detected. To investigate the deletion mechanism, we performed an in silico analysis of the genomic regions surrounding the breakpoints. No sequence homology between the breakpoint regions was found and hence, unequal homologous recombination seems unlikely to have caused the deletion. Accordingly, it would appear to be more probable that the deletion originated through a non-homologous recombination event mediated by several recombinogenic elements. In this regard, the 3′ breakpoint is at the border of a highly destabilized fragment of DNA with a high S/MAR potential that could be involved in generating the deletion, as reported previously.26, 27
With regards to the spectrum of SLC26A4 mutations in Spain, our results reveal a majority of missense mutations (69.2%). Even though this occurs in other populations, the four mutations most commonly detected among Caucasian patients are not very frequent (p.T416P and c.1001+1G>A) or do not appear in our Spanish population (p.E384G and p.L236P).12, 14 The mutational spectrum in Spain is similar to those of the Italian and French populations, sharing 12 of the SLC26A4 alleles with the French13 and four with the Italian population.15 However, in our population the most frequent mutation is the novel p.Q514K found in six alleles (17%) that derives from a common founder.
By means of a comprehensive genetic analysis, we have identified the first case of a de novo mutation c.1238A>C (p.Q413P) in the proband from a PS family that showed no apparent segregation with the DFNB4 locus. It is possible that the occurrence of de novo mutations may be the underlying reason for the large allelic heterogeneity of PS and non-syndromic EVA described in populations.
Regarding the prevalence of SLC26A4 mutations in our cohort, the presence of two mutations is significantly higher in PS (8/12, 60%) than in non-syndromic EVA (6/35, 17%) (P<0.009), as previously reported.10, 28 This difference was evident even though the group of non-syndromic EVA subjects may include some cases of PS. The difference would be even higher if the four PS patients with no SLC26A4 mutations are excluded from the count. These four patients aged between 8 and 17 years at time of clinical examination, did not show any thyroid alterations, and the discharge in the perchlorate test varied from 12–16%, values that should be considered as normal according to Pryor et al.10
In every PS proband and in the non-syndromic EVA patient E595.1 who carries only one mutated allele, we searched for mutations in both SLC26A4 exon 1 (containing the binding FOXI1 transcriptional regulatory elements) and the FOXI1 gene. No such mutations were found in any of these patients (data not shown). With respect to the incidence of ‘monoallelic’ cases, we found only one case (E595.1) who had a unilateral affectation (1/49, 2%, taking into account only the PS and non-syndromic EVA cases). In other groups studied, the incidence of ‘monoallelic’ cases is much higher, even though very different values have been reported. In a cohort of 100 French non-syndromic EVA, 16% were ‘monoallelic’ cases,13 while in other Caucasian groups of non-syndromic EVA or PS/non-syndromic EVA, 44%10 and 19%28 ‘monoallelic’ cases were found respectively. To reach a conclusion about the pathogenesis of the single mutated SLC26A4 alleles, more exhaustive genetic screenings should be performed to detect other SLC26A4 mutations that might exist, such as intragenic deletions or mutations in the regulatory regions. Besides, functional studies will be necessary to assess beyond doubt the pathogenic nature of the missense changes.
Accession codes
References
Everett LA, Glaser B, Beck JC et al: Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Genet 1997; 17: 411–421.
Li XC, Everett LA, Lalwani AK et al: A mutation in PDS causes non-syndromic recessive deafness. Nat Genet 1998; 18: 215–217.
Fraser GR : Association of congenital deafness with goitre (Pendred's syndrome) a study of 207 families. Ann Hum Genet 1965; 28: 201–249.
Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP : The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 1999; 21: 440–443.
Royaux IE, Suzuki K, Mori A et al: Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 2000; 141: 839–845.
Wangemann P, Nakaya K, Wu T et al: Loss of cochlear HCO3− secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol 2007; 292: F1345–F1353.
Royaux IE, Wall SM, Karniski LP et al: Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 2001; 98: 4221–4226.
Park HJ, Shaukat S, Liu XZ et al: Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 2003; 40: 242–248.
Blons H, Feldmann D, Duval V et al: Screening of SLC26A4 (PDS) gene in Pendred's syndrome: a large spectrum of mutations in France and phenotypic heterogeneity. Clin Genet 2004; 66: 333–340.
Pryor SP, Madeo AC, Reynolds JC et al: SLC26A4/PDS genotype phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet 2005; 42: 159–165.
Campbell C, Cucci RA, Prasad S et al: Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mut 2001; 13: 403–411.
van Hauwe P, Everett LA, Coucke P : Two frequent missense mutations in Pendred syndrome. Hum Mol Genet 1998; 7: 1099–1104.
Albert S, Blons H, Jonard L et al: SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet 2006; 14: 773–779.
Coyle B, Reardon W, Herbrick JA et al: Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet 1998; 7: 1105–1112.
Fugazzola L, Cerutti N, Mannavola D et al: Differential diagnosis between Pendred and pseudo-Pendred syndromes: clinical, radiologic and molecular studies. Pediatr Res 2002; 51: 479–484.
Yang T, Vidarsson H, Rodrigo-Blomqvist S et al: Transcriptional control of SLC26A4 is involved in Pendred syndrome and non-syndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet 2007; 80: 1055–1063.
Abeysinghe SS, Chuzhanova N, Krawczak M et al: Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum Mutat 2003; 22: 229–244.
Usami S, Abe S, Weston MD et al: Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet 1999; 104: 188–192.
Cremers CW, Admiraal RJ, Huygen PL et al: Progressive hearing loss, hypoplasia of the cochlea and widened vestibular aqueducts are very common features in Pendred's syndrome. Int J Pediatr Otorhinolaryngol 1998; 45: 113–123.
Prasad S, Kolln KA, Cucci RA et al: Pendred syndrome and DFNB4-mutation screening of SLC26A4 by denaturing high-performance liquid chromatography and the identification of eleven novel mutations. Am J Med Genet 2004; 124A: 1–9.
Kopp P, Arseven OK, Sabacan L et al: Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred's syndrome associated with a novel mutation in de PDS gene. J Clin Endocrinol Metabol 1999; 84: 336–341.
Palos F, García-Rendueles M, Araujo-Vilar D et al: Pendred syndrome in two Galician families: insights into clinical phenotypes through cellular, genetic and molecular studies. J Clin Endocrin Metab 2007; 93: 267–277.
Arellano B, Pera A, Ramirez-Camacho R et al: Pendred's syndrome and non-syndromic DFNB4 deafness associated with the homozygous T410 M mutation in the SLC26A4 gene in siblings. Clin Genet 2005; 67: 438–440.
Shears D, Conlon H, Murakami T et al: Molecular heterogeneity in two families with auditory pigmentary syndromes: the role of neuroimaging and genetic analysis in deafness. Clin Genet 2004; 65: 384–389.
Hao H, Wu L, Feng Y et al: Molecular analysis of hearing loss associated with enlarged vestibular aqueduct in the mainland Chinese: a unique SLC26A4 mutation spectrum. J Hum Genet 2007; 52: 492–497.
Albrecht P, Bode J, Buiting K et al: Recurrent deletion of a region containing exon 24 of the RB1 gene caused by non-homologous recombination between a LINE-1HS and MER21B element. J Med Genet 2004; 41: e122.
Xie F, Wang X, Cooper DN et al: A novel Alu-mediated 61-kb deletion of the von Willebrand factor (VWF) gene whose breakpoints colocate with putative matrix attachment regions. Blood Cells Mol Dis 2006; 36: 385–391.
Azaiez H, Yang T, Prasad S et al: Genotype–phenotype correlations for SLC26A4-related deafness. Hum Genet 2007; 122: 451–457.
Acknowledgements
We are grateful to the patients and the clinicians who collaborated in this study. We additionally thank Dolores Telleria for assistance with the DHPLC analysis and sequencing. This study was supported by the Spanish Ministerio de Ciencia y Tecnología research project SAF 2002-03966 and by the FP6 Integrated Project EUROHEAR (LSHG-CT-2004-512063).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pera, A., Villamar, M., Viñuela, A. et al. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss.. Eur J Hum Genet 16, 888–896 (2008). https://doi.org/10.1038/ejhg.2008.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.30
Keywords
This article is cited by
-
Probability of high-risk genetic matching with oocyte and semen donors: complete gene analysis or genotyping test?
Journal of Assisted Reproduction and Genetics (2022)
-
NGS-based expanded carrier screening for genetic disorders in North Indian population reveals unexpected results – a pilot study
BMC Medical Genetics (2020)
-
Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects
BMC Medical Genetics (2018)
-
The R130S mutation significantly affects the function of prestin, the outer hair cell motor protein
Journal of Molecular Medicine (2016)
-
GJB2 as Well as SLC26A4 Gene Mutations are Prominent Causes for Congenital Deafness
Cell Biochemistry and Biophysics (2015)
