
Abstract
Mutations in the long-range limb-specific cis-regulator (ZRS) could cause ectopic shh gene expression and are responsible for preaxial polydactyly (PPD). In this study, we analyzed a large Chinese isolated autosomal dominant PPD pedigree. By fine mapping and haplotype construction, we located the linked region to a 1.7 cM interval between flanking markers D7S2465 and D7S2423 of chromosome 7q36. We directly sequenced the candidate loci in this linked region, including the coding regions of the five genes (HLXB9, LMBR1, NOM1, RNF32 and C7orf13), the regulatory element (ZRS) of shh, the whole intron 5 of LMBR1 which contained the ZRS, and 18 conserved noncoding sequences (CNSs). Interestingly, no pathogenic mutation was identified. By using real-time quantitative PCR (qPCR), we also excluded the ZRS duplication in this pedigree. Our results indicate that, at least, it is not the mutation in a functional gene, CNS region or duplication of ZRS that cause the phenotype of this pedigree. The etiology of this PPD family still remains unclear and the question whether another limb-specific regulatory element of shh gene exists in the noncoding region in this 1.7 cM interval remains open for future research.
Similar content being viewed by others

Introduction
Vertebrate limb development malformations could lead to both polydactyly (extra digits) and oligodactyly (too few digits).1 Polydactyly is the most frequently observed congenital malformation with incidence rate between 5 and 19 per 10 000 live births.2 Polydactyly can occur either as an isolated disorder or syndrome. Furthermore, polydactyly can be classified into preaxial polydactyly (PPD) and postaxial polydactyly (PAP) according to its anatomical location. PPD phenotype is highly variable within families and ranges from a simple addition of a phalanx in triphalangeal thumb to whole digit duplication and tibial aplasia. According to Temtamy and McKusick,3 PPD can be divided into four types: type I (PPD-I) (OMIM 174400) is the duplication of one or more of the skeletal components of a biphalangeal thumb; type II (PPD-II) (OMIM 174500) is the polydactyly of a triphalangeal thumb; type III (PPD-III) (OMIM 174600) is the polydactyly of an index finger; type IV (PPD-IV) (OMIM 174700) is the polysyndactyly.
In the developing limb, asymmetric expression of Sonic Hedgehog (shh) in the zone of polarizing activity (ZPA) in posterior mesoderm is required for proper anterior/posterior (A/P) limb patterning.4 But in animal models and PPD families, mutations do not alter the shh coding sequence, which suggested that abnormal ectopic expression of shh is sufficient to induce supernumerary digits.5, 6, 7 Several unrelated PPD families under early genetic analysis had refined the PPD locus to a 450 kb region on chromosome 7q36, which covered five genes: HLXB9, LMBR1, NOM1, RNF32 and C7orf13.8, 9, 10, 11, 12, 13 So far, all described PPD pedigrees were linked to this locus.8, 9, 10, 11, 12, 13, 14, 15, 16, 17 Moreover, other limb disorders, such as acheiropodia18 (OMIM 200500), complex polysyndactyly,14 acropectoral syndrome19 (OMIM 605967) and currarino syndrome20 (OMIM 176450), were also mapped to this locus, which demonstrated that gene or regulatory elements vital for limb development are located in this region. Subsequent study of a spontaneous balanced translocation breakpoint in a girl with PDD and a transgenic inserted Ssq mouse indicated that intron 5 of LMBR1 contained a shh limb-specific regulatory element.21 Subsequent comparative genomic analysis revealed that this shh regulatory element (ZRS) was conserved in tetrapod lineage and teleost fishes but was absent or deleted among limbless reptiles and amphibians.22 In recent years, different point mutations in ZRS have been reported in PPD/TPTPS/SD4 families and in mouse and cat mutants.9, 13, 22, 23, 24, 25
To investigate the etiology of a large isolated PPD pedigree, which is the first Chinese PPD-II/III pedigree for molecular study, we performed linkage analysis on this pedigree. By using a fine mapping and haplotype analysis, we located the affected region to 1.7 cM between markers D7S2465 and D7S2423, including the 450-kb region mentioned above. But mutation screen by DNA sequencing gave us an unexpected result: no pathogenic mutation was found in the reported ZRS of shh, and subsequent extended screen indicated that there was no pathogenic mutation in the adjacent coding genes and conserved noncoding sequences (CNSs) in this linked 1.7 cM interval. As duplication involving ZRS is reported to be new pathogenic mechanism underlying TPTPS and SD4,16, 17 we also performed quantitative PCR (qPCR) in this pedigree. Our result showed that such rearrangement was not present in this family. Up to this stage the unconserved regions in this interval have not been determined and the question whether there is another regulatory element in the unconserved noncoding regions of this linked 1.7 cM interval remains open for future research.
Subjects and methods
Subjects
In this study, we recruited a six-generation pedigree including 45 Chinese Han individuals (21 affected members and 24 normal relatives) from a small village in Zhejiang Province (Figure 1). Meanwhile, 20 normal healthy individuals were recruited as controls. The detailed clinical investigation was performed for all affected members and peripheral blood samples were obtained for DNA analysis. Written informed consent was obtained from every subject. Autosomal-dominant inheritance of the phenotype was ascertained by the presence of affected individuals in each of the six generations and with a male–male transmission. The phenotype showed a complete penetrance in this pedigree.

Pedigree of the family studied and haplotypes obtained with 9 microsatellite DNA markers on chromosome 7q36. Solid symbols represent affected individuals; open symbols represent unaffected individuals. The sequence of markers is from centromere to telomere. The affected haplotype is shown in the rectangles.
Clinical phenotype
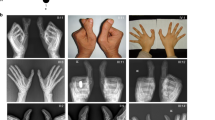
Total of 45 subjects were collected in this study, including 21 affected members, 12 healthy partners and 12 unaffected sibs. The affected members all have bilateral PPD and show hexadactyly of hands and feet (Figure 2). On the basis of physical examination, patients have the duplication of two phalanges or three phalanges thumb and show the phenotype of opposable thumb or non-opposable thumb. All affected members present a strikingly similar phenotype on feet with six fully developed toes. The supernumerary ray is localized between the first and the second ray. On the X-ray, the extra toe is biphalangeal and has the appearance of a somewhat hypolastic (duplicated) hallux (Figure 2b). No other abnormal of hand or feet and associated systemic anomalies are observed in this family. Therefore, we classified the clinical character of the family as isolated PPD-II/III according to Temtamy and McKusick's classification.3

Clinical phenotypes of affected individuals. (a) Normal images of hands and feet of affected V-37, which showed the hexadactyly. (b) The radiograms image of VI-47 shows a bilateral duplication of triphalangeal thumbs and the biphalangeal great toes. (c) Normal images of affected V-39 and V-11 showed the opposable thumb and non-opposable thumb.
Genotyping
The genomic DNA was extracted from peripheral blood by standard phenol/chloroform method. Nine microsatellite markers: D7S550, D7S3037, D7S2465, CGR13, CGR12, HING1, D7S559, CGR16 and D7S2423 were genotyped. The primers information was obtained from the Genome Database (GDB), the Généthon and the Cooperative Human Linkage Center collection (http://www.ncbi.nlm.nih.gov/sites/entrez?db=genome; http://www.genethon.fr/; http://www.chlc.org/) and the report from Heus et al.12 PCR was performed in a 15 μl volume with 10 ng genomic DNA as template, 1.5 μl PCR 10 × buffer, 1.5 μl dNTP mix (2.5 mM), 1 μl primers, 1 μl MgCl2 (15 mM), 0.12 U of Taq DNA polymerase (Takara, Dalian, China) and 9 μl DW water. Thermal cycling in a Gene Amp 9700 (PerkinElmer, Waltham, MA, USA) thermal cycler was performed at 95 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, followed by a 10 min extension at 72 °C and a hold at 10 °C. Then the PCR products were denatured and size fractionated on 5% denatured polyacrylamide gel (PAGE) run on ABI 377 sequencer. GS350 size standard was used as internal standard run in the same lane with the markers. Alleles read and scored by use of Genescan and Genotyper 2.0 software (ABI PerkinElmer, Waltham, MA, USA) were confirmed by eye.
Linkage analysis and haplotype construction
Two-point LOD scores were calculated by using MLINK program of Linkage software package (version 5.2). Linkage was determined in an autosomal-dominant model under penetrance of 95%. The gene frequency of disease was set to 0.001 and the mutation rate was set to zero. The recombination frequencies between male and female were assumed equal. Allele frequencies for all markers were kept equal. Haplotypes were constructed with software Simwalk2 (www.genetics.ucla.edu).
Sequence analysis of candidate regions
The candidate regions were amplified with genomic DNA. The conditions of PCR were identical to those mentioned above. PCR products were purified on spin columns (Watson Biotechnologies Inc., Shanghai, China) and were directly sequenced for both strands using BigDye Terminator Cycle Sequence Kit 3.1 (ABI Applied Biosystems, Foster City, CA, USA) according to the manufacturer's manual. The sequencing results were processed by DNASTAR software (DNASTAR) and manual rectification.
Real-time quantitative PCR
qPCR was performed on ABI Prism 7000 Sequence Detection System in a total volume of 20 μl in each well containing 10 μl of SYBR Premix Ex Taq (Takara), 1 μl of genomic DNA (20 ng), and 5 μl of Primers (400 nM each) with five replicates per sample. PCR conditions followed the manufacturer's protocol with 95 °C for 10 min and 40 cycles of 95 °C 15 s/60 °C 1 min. By using calibrator samples of normal control genomic DNA, the quantification of the target genes was normalized to an assay from chromosome 21 and the relative copy number was determined based on the ΔΔCt method.26 The ZRS was detected with primers previously described.16 The experiments were repeated five times and a conservative log2 ratio threshold of 0.3 and −0.3 was used for copy number gains or loses.
Results
Linkage analysis
A maximum two-point LOD score (Zmax) of 7.822 was obtained at recombination fraction (θ) 0.00 for marker HING1 within the LMBR1 gene (Table 1), which implied strong linkage between PPD phenotype of this kindred and chromosome 7q36. Haplotype construction and recombination analysis narrowed the candidate region to a 1.7 cM region between D7S2465 and D7S2423, which covered the previous reported 450-kb interval (Figure 1).12 Five genes (HLXB9, LMBR1, NOM1, RNF32 and C7orf13), as well as the regulatory element ZRS within the intron 5 of LMBR1, were located inside this 1.7 cM interval.8, 9, 10, 11, 12, 13
Mutation screen of candidate genes
We performed a comprehensive mutation screening in the coding regions of HLXB9, LMBR1, NOM1, RNF32 and C7orf13. No sequence alterations of the amino acid coding of these five genes were found. Then the 3′-untranslated regions and their intron–exon boundaries of the five genes, as well as the cis-acting regulator of shh (ZRS), were also screened and no pathogenic mutation was found. In addition, no deleterious sequence changes were found in all the alternative splicing transcripts of the five genes and three annotated genes (Loc100129517, Loc100129551 and Loc645249) in the region.
Then, the whole intron 5 of LMBR1 containing the ZRS was sequenced. A total of 18 single-nucleotide polymorphisms (SNPs) were found (Table 2) but none of them was pathogenic mutation. Furthermore, all of them were also detected in normal healthy controls. These results showed that there is no deleterious mutation in the coding regions and reported regulatory element (ZRS) of this 1.7 cM interval in this pedigree.
As reported in families of acheiropody, a severe disorder presenting with bilateral amputations of upper and lower extremities, a homozygous deletion of 4–6 kb containing LMBR1 exon 4 was the etiology. We also scanned the ∼7 kb region of acheiropody deletion in our pedigree, but still no pathogenic mutation was identified.
Identification and sequencing of the conserved noncoding sequence
Highly CNSs among diverse species have been predicted to be involved in regulation of transcription, DNA replication, chromosome pairing and chromosome condensation.27 Recently, CNS was also suggested to be associated with vertebrate development28 and in fact, the ZRS of shh was highly conserved in tetrapod lineage and even in teleost fishes.22 So we sequenced CNSs within this 1.7 cM interval.
The linked 1.7 cM region of human and its homologous sequences from several widely divergent organisms (mouse, rat, dog and chick) were downloaded from UCSC (http://genome.ucsc.edu/; human (chromosome7: 156 033 000–156 493 000 Mar 2006, UCSC hg18); mouse (July 2007, UCSC mm9); rat (Nov 2004, UCSC rn4); dog (May 2005); chicken (Feb 2004, UCSC version gal2)). The multiple species sequences were aligned by VISTA (http://www-gsd.lbl.gov/vista) and displayed using PileUP (The GCG Wisconsin Package at the HGMP, Hinxton, UK).29 All sequences were soft-masked and aligned using SLAGAN alignment tool kit (http://www-gsd.lbl.gov/vista).
A total of 18 CNSs shared by human–mouse, human–rat, human–dog and human–chicken alignment were found and subsequently sequenced (Table 3). After sequencing analysis, we detected 10 SNPs among the 18 CNSs and all the 10 SNPs were also present in normal healthy controls. Therefore, they could not be pathogenic mutations accounting for PPD.
Identification of the ZRS duplication
ZRS duplication was found in some families of TPTPS and SD4 phenotype.16, 17 qPCR was used to detect whether the copy number changes of ZRS was present in this PPD pedigree. We examined 8 affected individuals, 8 normal individuals from the family and 9 unrelated healthy Han Chinese controls with the same ethnic background. The duplication of ZRS was not detected and the mean relative copy number was 1.0 (mean value 0.95–1.08) in the affected group.
Discussion
It is not easy to choose proper organisms for interspecies sequence comparison of a particular DNA region due to the inconsistent mutation rates across the genome,30 which might be one of the factors contributing to our negative result. Another limitation for multispecies sequence comparison may overlook the regulatory element whose sequence or position in the genomes has not been conserved across phyla, such as some enhancers.31
In previous PPD studies, there are only 13 pedigrees in which the responsible point mutations were identified.13, 16, 17, 23, 25 On the other hand, pathogenic mutations are still remained unidentified in most PPD families.8, 11, 12, 13, 23 Furthermore, although the 7 pathogenic point mutations identified in above 13 pedigrees were all located in the ZRS of shh, these mutation sites were scattered in the ZRS and no clustering of mutations toward domain-like structure was evident. In fact, no known regulatory domain had been defined by bioinformatics method, so far.13 These information suggested that the network of shh regulation was much complex and is poorly understood.
Recently, the copy number variations, such as duplication, has been shown to be the underlying cause for human genetic disorders.32 As shh gene is expressed in different tissues and developmental stages, more accurate and multiple complex regulators are necessary. Very recently, two different research teams have found a genomic duplication fragment containing ZRS in TPTPS and SD4 families,16, 17 but the precise molecular mechanism about how the duplication cause human limb disorders is unclear. The phenotype of TPTPS and SD4 are more severe and complicated than isolated PPD, which cannot be simply explained by shh ectopic expression, though all the disorders are confined to human limbs. Duplication of ZRS may have more severe deleterious effect on shh expression than single-nucleotide alterations in the ZRS. As all affected individuals in our PPD kindred only had the simple phenotype of bilateral hexadactyly hand and foot without any other features, the presumed mutation in this PPD family should be one causing the disruption of distal A/P axis formation and mildly dysregulate shh expression. qPCR was used to detect the duplication of ZRS and showed that ZRS rearrangement was not the pathogenic mutation in our pedigree, which was consistent with this postulation based on the phenotype.
The question whether there is other regulatory element of shh in the defined 1.7 cM interval, in addition to the known ZRS, is intriguing. The studies of de novo reciprocal t(5,7)(q11,136) translocation and the acheiropody family with 4–6 kb deletion containing LMBR1 exon 4 implied the presence of other limb-specific regulator of shh expression.18, 21 In our pedigree, the fact that all the coding regions and conserved regions were excluded from containing pathogenic mutation suggests that the other regulatory element could, if it exists, locate in unconserved regions. It seems likely that the only way to detect the proposed pathogenic mutation for our pedigree is to thoroughly sequence the remained unconserved regions in this 1.7 cM interval. The eventual characterization of the pathogenic mutation for this family could help better understand the complex developmental gene regulatory circuitry in human limb embryogenesis.
References
Clark RM, Marker PC, Roessler E et al: Reciprocal mouse and human limb phenotypes caused by gain- and loss-of-function mutations affecting Lmbr1. Genetics 2001; 159: 715–726.
Castilla E, Paz J, Mutchinick O, Munoz E, Giorgiutti E, Gelman Z : Polydactyly: a genetic study in South America. Am J Hum Genet 1973; 25: 405–412.
Temtamy SA, McKusick VA : The genetics of hand malformations. Birth Defects Orig Artic Ser 1978; 14: i–xviii, 1–619.
Mariani FV, Martin GR : Deciphering skeletal patterning: clues from the limb. Nature 2003; 423: 319–325.
Clark RM, Marker PC, Kingsley DM : A novel candidate gene for mouse and human preaxial polydactyly with altered expression in limbs of Hemimelic extra-toes mutant mice. Genomics 2000; 67: 19–27.
Hill RE, Heaney SJ, Lettice LA : Sonic hedgehog: restricted expression and limb dysmorphologies. J Anat 2003; 202: 13–20.
Knudsen TB, Kochhar DM : The role of morphogenetic cell death during abnormal limb-bud outgrowth in mice heterozygous for the dominant mutation Hemimelia-extra toe (Hmx). J Embryol Exp Morphol 1981; 65 Suppl: 289–307.
Zguricas J, Heus H, Morales-Peralta E et al: Clinical and genetic studies on 12 preaxial polydactyly families and refinement of the localisation of the gene responsible to a 1.9 cM region on chromosome 7q36. J Med Genet 1999; 36: 32–40.
Wang ZQ, Tian SH, Shi YZ et al: A single C to T transition in intron 5 of LMBR1 gene is associated with triphalangeal thumb-polysyndactyly syndrome in a Chinese family. Biochem Biophys Res Commun 2007; 355: 312–317.
Hing AV, Helms C, Slaugh R et al: Linkage of preaxial polydactyly type 2 to 7q36. Am J Med Genet 1995; 58: 128–135.
Heutink P, Zguricas J, van Oosterhout L et al: The gene for triphalangeal thumb maps to the subtelomeric region of chromosome 7q. Nat Genet 1994; 6: 287–292.
Heus HC, Hing A, van Baren MJ et al: A physical and transcriptional map of the preaxial polydactyly locus on chromosome 7q36. Genomics 1999; 57: 342–351.
Gurnett CA, Bowcock AM, Dietz FR, Morcuende JA, Murray JC, Dobbs MB : Two novel point mutations in the long-range SHH enhancer in three families with triphalangeal thumb and preaxial polydactyly. Am J Med Genet A 2007; 143: 27–32.
Tsukurov O, Boehmer A, Flynn J et al: A complex bilateral polysyndactyly disease locus maps to chromosome 7q36. Nat Genet 1994; 6: 282–286.
Zguricas J, Snijders PJ, Hovius SE, Heutink P, Oostra BA, Lindhout D : Phenotypic analysis of triphalangeal thumb and associated hand malformations. J Med Genet 1994; 31: 462–467.
Sun M, Ma F, Zeng X et al: Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long-range, limb-specific SHH enhancer. J Med Genet 2008; 45: 589–595.
Klopocki E, Ott CE, Benatar N, Ullmann R, Mundlos S, Lehmann K : A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J Med Genet 2008; 45: 370–375.
Ianakiev P, van Baren MJ, Daly MJ et al: Acheiropodia is caused by a genomic deletion in C7orf2, the human orthologue of the Lmbr1 gene. Am J Hum Genet 2001; 68: 38–45.
Dundar M, Gordon TM, Ozyazgan I et al: A novel acropectoral syndrome maps to chromosome 7q36. J Med Genet 2001; 38: 304–309.
Ross AJ, Ruiz-Perez V, Wang Y et al: A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet 1998; 20: 358–361.
Lettice LA, Horikoshi T, Heaney SJ et al: Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc Natl Acad Sci USA 2002; 99: 7548–7553.
Sagai T, Masuya H, Tamura M et al: Phylogenetic conservation of a limb-specific, cis-acting regulator of Sonic hedgehog (Shh). Mamm Genome 2004; 15: 23–34.
Lettice LA, Heaney SJ, Purdie LA et al: A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 2003; 12: 1725–1735.
Masuya H, Sezutsu H, Sakuraba Y et al: A series of ENU-induced single-base substitutions in a long-range cis-element altering Sonic hedgehog expression in the developing mouse limb bud. Genomics 2006.
Lettice LA, Hill AE, Devenney PS, Hill RE : Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum Mol Genet 2008; 17: 978–985.
Lyle R, Radhakrishna U, Blouin JL et al: Split-hand/split-foot malformation 3 (SHFM3) at 10q24, development of rapid diagnostic methods and gene expression from the region. Am J Med Genet A 2006; 140: 1384–1395.
Nobrega MA, Pennacchio LA : Comparative genomic analysis as a tool for biological discovery. J Physiol 2004; 554: 31–39.
Bejerano G, Pheasant M, Makunin I et al: Ultraconserved elements in the human genome. Science 2004; 304: 1321–1325.
Mayor C, Brudno M, Schwartz JR et al: VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000; 16: 1046–1047.
Woolfe A, Goodson M, Goode DK et al: Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol 2005; 3: e7.
Frazer KA, Tao H, Osoegawa K et al: Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res 2004; 14: 367–372.
Beckmann JS, Estivill X, Antonarakis SE : Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 2007; 8: 639–646.
Acknowledgements
We are grateful to the family members for participating in this research. We thank Professor Nelson Leung-Sang Tang and Dr Huai-Rong Luo for their help in preparing the manuscript. This work was supported by the Key State Research Program of China (2006CB943900), Science and Technology Committee of Yunnan Province and the National Natural Science Foundation of China (30621092, 30671162).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, H., Wang, CY., Wang, JX. et al. Mutation analysis of a large Chinese pedigree with congenital preaxial polydactyly. Eur J Hum Genet 17, 604–610 (2009). https://doi.org/10.1038/ejhg.2008.240
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.240
Keywords
This article is cited by
-
The molecular genetics of human appendicular skeleton
Molecular Genetics and Genomics (2022)
-
Identity-by-descent refines mapping of candidate regions for preaxial polydactyly II /III in a large Chinese pedigree
Hereditas (2018)
-
The disruption of a novel limb cis-regulatory element of SHH is associated with autosomal dominant preaxial polydactyly-hypertrichosis
European Journal of Human Genetics (2016)
