
Abstract
Cystic fibrosis (CF) is an autosomal recessive disease that results from mutations in the CF transmembrane conductance regulator (CFTR) gene. The effect of interventions aimed at correcting the CF electrophysiologic phenotype has been primarily measured using in vitro methods in gastrointestinal and respiratory epithelia. A reliable in vivo assay of CFTR function would be of great value in the investigation of pharmacologic interventions for CF mouse models. We performed the in vivo rectal potential difference (RPD) assay on three different mouse models. We then compared the in vivo data with the results obtained using the in vitro Ussing chamber method. The results from the in vitro method correlated closely with the results acquired using the in vivo method and were reproducible. The data suggest that the in vivo RPD assay is a reliable assay of functional CFTR expression in CF mouse models.
Similar content being viewed by others


Main
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, which codes for the cystic fibrosis transmembrane conductance regulator (CFTR) (1–3). CFTR, a member of the adenosine triphosphate binding cassette transporter glycoprotein superfamily, functions as a chloride channel and regulates other ion channels in the apical membrane of certain epithelia. Over a thousand disease-causing mutations have been identified in the CFTR gene. These mutations have been separated into five major subtypes, which include null/decreased production (Class I), improper processing and trafficking (Class II), ion channel dysregulation (Class III), altered channel conduction (Class IV), and improper protein targeting (Class V). The most common mutation in the CFTR gene is ΔF508, a Class II mutation in which the CFTR protein is retained in the endoplasmic reticulum and targeted for degradation (4–6). ΔF508-CFTR can function as a chloride channel but is unable to reach the apical membrane (7,8).
A number of future therapeutic strategies for CF are focused on correcting or circumventing the mutant CFTR gene, with the hope that this will result in decreased pathology caused by the primary gene defect. A variety of compounds are being investigated as potential treatments in CF, for instance, 4-phenylbutyrate, trimethylamine oxide (TMAO), and curcumin, as well as flavonoids. 4-Phenylbutyrate, TMAO, and curcumin have been shown to increase ΔF508-CFTR surface expression in a number of in vitro systems (9–11), whereas once at the membrane, flavonoid compounds (genistein, apigenin) and vitamin C have been suggested to activate ΔF508-CFTR (12–14). In addition, gentamicin, an aminoglycoside antibiotic, has been shown to suppress premature termination codons and restore CFTR function in patients with the W1282X mutation (15). Other interventions, such as gene therapy using viral vectors and stem cell transplant, have also been suggested as potential interventions in CF.
Assessing the effects of the various interventions aimed at correcting the CF electrophysiologic phenotype in affected epithelia is primarily performed using in vitro electrophysiologic methods such as examining excised tissue mounted in Ussing chambers. The nasal potential difference (PD) assay is a widely accepted in vivo method, which has been used to assess functional expression of CFTR in respiratory epithelia in humans and was pioneered by Knowles et al. (16,17) in the 1980s. This method has been adapted by Grubb (18) and now allows for the investigation of many potential interventions for CF in various mouse models of CF. Similarly, a reliable in vivo assay of CFTR function in intestinal epithelia could be of great value in the investigation of pharmacologic interventions for CF mouse models. It has been suggested that the rectal potential difference (RPD) assay is a reliable in vivo method for assessment of CFTR activity (10, 11, 19). However, there are no reported studies that evaluate the reliability and reproducibility of this method.
METHODS
Mice.
Two models of gene-targeted mice for the ΔF508 mutation were used in the experiments. One of the mouse models was bred on a mixed genetic background [approximately 25% 129 and 75% Black 6 (Bl6) (20)], whereas the other ΔF508 mouse model was further backcrossed into a pure Bl6 background. We refer to these mouse models respectively as the mixed genetic background and pure Bl6 background ΔF508 mice. A transgenic CFTR−/− knockout (B6.129P2Cftrtm1Unc) was also used in the experiments as a CFTR-null model (21,22). The ΔF508-CFTR+/+ and the CFTR-null mice have severe gastrointestinal manifestations, including bowel obstruction, bowel strictures, and peritonitis. All mice were fed with Harlan Teklad 9F food (Harlan Teklad, Madison, WI) and drinking water that was supplemented with 17.5 g/250 mL of Colyte (Schwarz Pharma, Milwaukee, WI) to improve the survival of the animals (23). All three CF mouse models were bred to obtain wild-type, heterozygote, and CF-affected animals for each model system, were maintained at the Yale University Animal Facility, and were genotyped with standard protocols. All procedures were performed in compliance with relevant laws and institutional guidelines and were approved by the Yale University Institutional Animal Care and Use Committee.
In vivo potential difference measurements.
The intestinal PD was measured using a probing bridge made from PE50 tubing inserted into the rectum of the mouse. A 3 mm insertion depth for RPD and a 15 mm insertion depth for “distal colon” PD (DCPD) were used. AgCl2 voltage electrodes were connected by way of 3% agar bridges made with 3 M KCl. The insertion depths for the rectal and distal colon have been confirmed at the time of sacrifice to ensure proper placement of the tubing. The experimental electrode was connected through the probing bridge, and the reference electrode was connected through a 23-gauge butterfly needle filled with normal saline and placed subcutaneously. A microperfusion pump controlled the flow of perfusate through the probing electrode (0.5 mL/h). A separate length of PE50 tubing was inserted in the rectum and attached to a suction apparatus to minimize distension of the gut. During the procedure, the mice were maintained on a warming pad, and saline drops or artificial tears were used to keep their eyes moist. Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) intraperitoneally before the procedure. Mice were continually observed throughout the procedure and postprocedure until they were fully recovered.
In vitro potential difference measurements.
We performed the in vitro analysis as previously described by Grubb (18). The rectum and distal colon were removed, cut longitudinally, washed of any intestinal contents, and mounted in the Ussing chamber with an aperture size of 0.04 cm2 for rectal tissue and 0.3 cm2 for distal colonic tissue (Physiologic Instruments, San Diego, CA). The PD was measured under open-circuit conditions. During all experiments, solutions were continuously gassed with 95% O2/5% CO2 and warmed to 37°C. Data were recorded continuously using the Acquire and Analyze software program (Physiologic Instruments, San Diego, CA).
Solutions.
The solutions used in this study were a Kreb's bicarbonate Ringer's solution (KBR) and a chloride-free Ringer's solution containing 5 mM barium hydroxide. Amiloride and forskolin were added to the chloride-free Ringer's solution at final concentrations of 10−4 M and 10−5 M, respectively. In a subset of experiments, a specific CFTR inhibitor, CFTRinh-172, was added to the final perfusate. All drugs and chemicals were obtained from Sigma Chemical Co. (St. Louis, MO) and J. T. Baker (Phillipsburg, NJ).
Statistics.
The t test was used to compare groups for significant differences. A p value of less than 0.05 was used to signify statistical significance. All results are reported as the mean ± SEM.
RESULTS
In vivo intestinal potential difference.
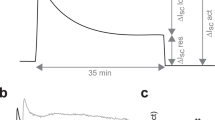
The in vivo intestinal PD was measured for 30 min. Initially, a KBR solution is perfused for a baseline measurement. It is quickly switched to the first perfusate solution that is used in the RPD assay, a chloride-free Ringer's solution containing Ba(OH)−2 and amiloride, as previously described by Fischer et al. (10,14), and the transepithelial PD is recorded for 15 min. The addition of amiloride and barium block sodium and potassium currents that could contribute to the PD. Subsequently, forskolin was added to the perfusate, and the PD was measured for an additional 15 min to activate CFTR chloride current. Data were obtained from two separate regions of the murine intestinal tract, the distal colon and the rectum (Fig. 1). The baseline PD, the mean change in the PD in response to forskolin (ΔPDfsk), and the peak forskolin PD for all CF mouse models were obtained for distal colon (Table 1) and rectum (Table 2).

Representative tracings of in vivo DCPD. Intestinal PD was measured using a probing bridge made from PE50 tubing inserted 15 mm in depth, as outlined in the “Methods” section. During the initial period, the PD is measured in a chloride-free Ringer's solution containing amiloride and barium hydroxide. Subsequently, forskolin is added to the perfusing solution. (A) Representative tracing from a nonCF animal. (B) CF-affected mouse.
At baseline, in vivo intestinal PD measurements of wild-type and heterozygote mice revealed a modest hyperpolarization, which was similar in magnitude for all three models. Furthermore, in wild-type and heterozygous mice, the addition of forskolin resulted in a further hyperpolarization (a more negative PD measurement). This observation held true in all three models for both the distal colon and rectum (Tables 1 and 2). Interestingly, in both distal colon and rectum, the wild-type mice demonstrated a more negative baseline PD, a more negative peak PD in response to forskolin, and a larger ΔPDfsk than the heterozygous mice (Tables 1 and 2). However, these trends did not reach statistical significance except in the CFTR-null mouse model.
The baseline PD in CF-affected mice was significantly more positive when compared with the heterozygous and wild-type mice. This observation held true for all three CF mouse models regardless of the intestinal region studied (Tables 1 and 2). In addition, whereas all of the wild-type and heterozygous mice demonstrated a hyperpolarization of the PD in response to forskolin, the CF-affected mice demonstrated a depolarization of the PD in response to forskolin (Tables 1 and 2). These differences are similar to previously reported in vivo findings and are consistent with the CF phenotype (10).
To demonstrate that the forskolin stimulated current was caused by CFTR activity, 20 μM of CFTRinh-172, a specific CFTR inhibitor, was added to the perfusate solution after forskolin stimulation in a subset of experiments (24). The addition of the blocker significantly inhibited the hyperpolarization that was observed in the wild-type and heterozygote animals in all three mouse models (n = 16). These findings suggest that the majority of the current activated by forskolin was caused by CFTR activation. No effect was observed in the CF-affected animals (n = 6) (Fig. 2).

CFTRinh-172 inhibits the forskolin stimulated hyperpolarization observed in the in vivo RPD/DCPD assay in all three CF animal models. Twenty micromolar of CFTRinh-172 was added to the chloride-free Ringer's solution containing amiloride, barium hydroxide, and forskolin. Mixed background ΔF508-CFTR colony (n = 6); CFTR null colony (n = 5); pure Bl6 background ΔF508-CFTR colony (n = 5).
In vivo ΔPDfsk is a reproducible measurement when assayed repeatedly on a single animal.
To test the reproducibility of the in vivo methods, we repeated three in vivo DCPD assays on five heterozygous and six CF-affected mice at 2 wk to monthly intervals. Although there is some variability in the absolute values, the ΔPDfsk remained similar for all of three experiments for each animal (Fig. 3) For instance, the ΔPDfsk for one of the heterozygous mice was −10.7, −11.2, and −9.6 mV, respectively, and for one of the CFTR-null mice was 3.3, 2.0, and 2.5 mV, as noted in Fig. 3.

In vivo PD measurements in CF-affected and normal mice are reproducible. Repeated DCPD measurements were obtained in three separate experiments on one heterozygote (CFTR null mouse) and on one homozygote CFTR null mouse. (A) PD measurements for a heterozygous (CFTR-null) mouse are graphed over time: trial #1 (♦), trial #2 (▪), trial #3 (▴), and mean values (○) for the three experiments. Horizontal black bar represents a time length of 15 min. Black arrow denotes change to forskolin containing solution. (B) ΔPDfsk for each trial depicted in A: trial #1 (□), trial #2 (▪), trial #3 ( ), and the mean (
), and the mean ( ) ΔPDfsk for the three experiments. (C) The average ΔPDfsk for three repeated RPD assays on five heterozygote mice. (D) PD measurements for a homozygous (CFTR-null) mouse are graphed over time: trial #1 (♦), trial #2 (▪), trial #3 (▴), and the mean values (○) for the three experiments. Horizontal black bar represents a time length of 15 min. Black arrow denotes change to forskolin containing solution. (E) ΔPDfsk for each trial is depicted in D: trial #1 (□), trial #2 (▪), trial #3 (), and the mean () ΔPDfsk for the three experiments. (F) The average ΔPDfsk for three repeated RPD assays on six CF-affected mice. In all panels, error bars represent the SEM.
) ΔPDfsk for the three experiments. (C) The average ΔPDfsk for three repeated RPD assays on five heterozygote mice. (D) PD measurements for a homozygous (CFTR-null) mouse are graphed over time: trial #1 (♦), trial #2 (▪), trial #3 (▴), and the mean values (○) for the three experiments. Horizontal black bar represents a time length of 15 min. Black arrow denotes change to forskolin containing solution. (E) ΔPDfsk for each trial is depicted in D: trial #1 (□), trial #2 (▪), trial #3 (), and the mean () ΔPDfsk for the three experiments. (F) The average ΔPDfsk for three repeated RPD assays on six CF-affected mice. In all panels, error bars represent the SEM.
In Vivo Intestinal Potential Difference versus In Vitro Intestinal Potential Difference.
Next, to test whether the in vivo PD assay results correlated with those observed in an in vitro assay, we measured the in vitro intestinal PD using the Ussing chamber. Before the in vitro assay, in vivo PD measurements were obtained on each animal. Immediately after the in vivo assay, the animals were killed, and the in vitro assay was performed. Data were again obtained from two separate regions of the murine intestinal tract, the distal colon and the rectum, after a 20-min equilibration period in KBR. The same protocol and solutions were used in the Ussing chamber as in the in vivo PD method. The mean baseline RPD and DCPD were similar in all three mouse models regardless of whether the Ussing chamber or the RPD method was used to obtain measurements (Figs. 4 and 5).

Comparison of the observed change in PD in response to 10 μM forskolin (ΔPDfsk) in wild-type (□), heterozygousL (), and CF-affected mice (▪) in three CF mouse models. In vitro results are represented on the left side of each panel and in vivo results on the right side. n = 5 for all groups (wild type, heterozygote, CF affected) and both tissues (rectum and distal colon). *p < 0.05 between in vivo and in vitro results for distal colon in CF-affected groups only. (A) ΔF508-CFTR mixed background mouse model. (B) CFTR-null CF mouse model. (C) ΔF508-CFTR pure Bl6 background mouse model.

Comparison of the mean PD measured at minute intervals using the in vitro assay (closed symbols) or the in vivo (open symbols) assay. CF results are represented as circles, wild type as squares, and heterozygote as triangles. Horizontal black bar represents a time length of 15 min. Black arrow denotes change to forskolin containing solution. (A and B) Traces for the mixed background ΔF508 mice (distal colon A, rectum B). (C and D) Traces for the CFTR-null mice (distal colon C, rectum D). (E and F) Traces from the pure Bl6 background ΔF508 mice (distal colon E, rectum F).
For all three CF mouse models, the forskolin response in both the distal colon and rectum appeared similar in magnitude when using either in vivo or in vitro methods to measure the ΔPDfsk (Figs. 4 and 5), and the inhibition of the forskolin response by CFTRinh-172 was also similar. There was only one significant difference observed between in vivo and in vitro measurements, and it was observed in the CF-affected mice. In the CF mice, there was a greater depolarization in the in vivo distal colon (ΔPDfsk) analysis compared with the in vitro results. This was true for all three CF mouse models. The distal colon ΔPDfsk measurements were 3.8 ± 0.4 mV versus. 0.9 ± 0.3 mV for mixed genetic background ΔF508 mouse model, 3.7 ± 0.3 mV versus 1.1 ± 0.2 mV for CFTR-null model, and 3.9 ± 0.6 mV versus 1.5 ± 0.3 for pure Bl6 background ΔF508 mouse model. A similar trend was also observed for the rectal PD of the CF mice; however, it was not of statistical significance. Last, a significant inhibition of the forskolin response was observed with the addition of CFTRinh-172 in both the wild-type and heterozygous tissues, similar to what observed in the in vivo assay (n = 9, data not shown). Once again, there was no effect of CFTRinh-172 on the CF-affected tissue (n = 5, data not shown).
DISCUSSION
The in vivo rectal PD assay appears to be a reliable and reproducible assay of functional CFTR expression in CF mouse models. In our study, the in vivo PD measurements correlated very closely with the PD measurements acquired using the in vitro method, suggesting that the in vivo PD assay may be a very useful alternative method for studying ion transport in CF-affected animals if serial measurements are required as part of a study design. Although the results of the two methods are nearly identical, there were interesting subtle differences when comparing techniques. Of note, in CF-affected animals, the degree of depolarization observed in response to forskolin appeared to be greater when using the in vivo assay than the in vitro assay.
In addition, in the in vivo PD assay, wild-type mice demonstrated a larger ΔPDfsk, a more negative baseline PD, and a more negative peak PD in response to forskolin compared with their heterozygote counterparts, suggesting that there is a greater degree of functional CFTR expression in wild-type than in heterozygous mice. This trend was noted for all three CF mouse models studied, in both the distal colon and rectum. Our in vivo data correlate with the observations of Gabriel et al. (25). They reported that heterozygote CFTR(−/+) mice were more resistant to secretory diarrhea after exposure to cholera toxin when compared with wild-type animals. They postulated that this heterozygote advantage was caused by less CFTR expression in the CFTR heterozygous mice (26). Interestingly, the PD measurements from the same mice using the in vitro method did not show statistically significant differences between the wild-type and heterozygous groups, which is similar to the findings of Cuthbert et al. (26) and is consistent the initial characterization of intestinal transport by in vitro methods in the ΔF508 mixed genetic background mouse model (19). We speculate that the subtle differences that are detected using the in vivo method result from the fact that the tissue is in situ and is subject to the endogenous neural-humoral supply of the gut. This is distinctly different from the removal and mounting of the same tissue in an artificial environment to maintain tissue viability.
Both methods of studying intestinal transport have their limitations. For example, serial measurements of CFTR functional expression are not possible when using the in vitro method. On the other hand, it is impossible to control the internal and external environments affecting the tissue when using the in vivo method. However, both methods used in concert can yield important details regarding the ion transport properties of the intestinal epithelia. The power of the in vivo method is its ability to measure CFTR functional expression before and after interventions aimed at correcting the CF electrophysiologic phenotype.
Abbreviations
- Bl6:
-
Black 6
- CF:
-
cystic fibrosis
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- CFTR(−/+):
-
heterozygous for wild type CFTR and mutant CFTR
- CFTRinh-172:
-
a specific CFTR channel inhibitor
- DCPD:
-
distal colon potential difference
- ΔF508:
-
deletion of phenylalanine at the 508 position
- ΔPDfsk:
-
mean change in potential difference in response to forskolin
- KBR:
-
Kreb's bicarbonate Ringer's solution
- PD:
-
potential difference
- RPD:
-
rectal potential difference
- TMAO:
-
trimethylamine oxide
References
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui L 1989 Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073
Davis PB, Drumm M, Konstan MW 1996 Cystic fibrosis. Am J Respir Crit Care Med 154: 1229–1256
Sheppard DN, Welsh MJ Structure and function of the CFTR chloride channel. Physiol Rev 1999 79: S23–S45
Thomas PJ, Shenbagamurthi P, Sondek J, Hullihen JM, Pedersen PL 1992 The cystic fibrosis transmembrane conductance regulator. Effects of the most common cystic fibrosis-causing mutation on the secondary structure and stability of a synthetic peptide. J Biol Chem 267: 5727–5730
Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE 1990 Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834
Ward CL, Omura S, Kopito RR 1995 Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83: 121–127
Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ 1992 Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764
Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS 1991 Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 254: 1797–1799
Rubenstein RC, Egan ME, Zeitlin PL 1997 In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 100: 2457–2465
Fischer H, Fukuda N, Barbry P, Illek B, Sartori C, Matthay MA 2001 Partial restoration of defective chloride conductance in DeltaF508 CF mice by trimethylamine oxide. Am J Physiol Lung Cell Mol Physiol 281: L52–L57
Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ 2004 Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304: 600–602
Illek B, Fischer H, Santos GF, Widdicombe JH, Machen TE, Reenstra WW 1995 cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am J Physiol 268: C886–C893
Illek B, Fischer H 1998 Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. Am J Physiol 275: L902–L910
Fischer H, Schwarzer C, Illek B 2004 Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A 101: 3691–3696
Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E 2003 Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 349: 1433–1441
Knowles M, Gatzy J, Boucher R 1981 Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 305: 1489–1495
Knowles M, Gatzy J, Boucher R 1983 Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest 71: 1410–1417
Grubb BR 2002 Bioelectric measurement of CFTR function in mice. In: Skach WR (ed) Methods in Molecular Medicine. Humana Press, Inc., Totowa, NJ, pp 525–535
Wilschanski MA, Rozmahel R, Beharry S, Kent G, Li C, Tsui LC, Durie P, Bear CE 1996 In vivo measurements of ion transport in long-living CF mice. Biochem Biophys Res Commun 219: 753–759
Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Capecchi MR, Welsh MJ, Thomas KR 1995 A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest 96: 2051–2064
Koller BH, Kim HS, Latour AM, Brigman K, Boucher RC Jr, Scambler P, Wainwright B, Smithies O 1991 Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. Proc Natl Acad Sci U S A 88: 10730–10734
Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH 1992 An animal model for cystic fibrosis made by gene targeting. Science 257: 1083–1088
Clarke LL, Gawenis LR, Franklin CL, Harline MC 1996 Increased survival of CFTR knockout mice with an oral osmotic laxative. Lab Anim Sci 46: 612–618
Taddei A, Folli C, Zeggra-Moran O, Fanen P, Verkman AS, Gailetta LJ 2004 Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett 558: 52–56
Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ 1994 Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266: 107–109
Cuthbert AW, Halsted J, Ratcliff R, Colledge WH, Evans MJ 1995 The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol 482: 449–454
Acknowledgements
We would like to acknowledge Dr. Horst Fischer for his extremely helpful communications during the development of our in vivo rectal potential difference assay. We would also like to acknowledge and thank Jeffrey Wisner for his extraordinary technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by grants from the National Institutes of Health (DK53428, MEE) and the Cystic Fibrosis Foundation (EGAN04G0, MEE.). S.A.W. is supported by a training grant from the National Institutes of Health (T32HD07094).
Rights and permissions
About this article
Cite this article
Weiner, S., Caputo, C., Bruscia, E. et al. Rectal Potential Difference and the Functional Expression of CFTR in the Gastrointestinal Epithelia in Cystic Fibrosis Mouse Models. Pediatr Res 63, 73–78 (2008). https://doi.org/10.1203/PDR.0b013e31815b4bc6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31815b4bc6
This article is cited by
-
The Ussing chamber system for measuring intestinal permeability in health and disease
BMC Gastroenterology (2019)
-
Colonic potassium handling
Pflügers Archiv - European Journal of Physiology (2010)
