- NEWS AND VIEWS
Organoids reveal cancer dynamics
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 556, 441-442 (2018)
doi: https://doi.org/10.1038/d41586-018-03841-x
References
Roerink, S. F. et al. Nature 556, 457–462 (2018).
Sottoriva, A. et al. Nature Genet. 47, 209–216 (2015).
Sun, R. et al. Nature Genet. 49, 1015–1024 (2017).
Vogelstein, B. et al. Science 339, 1546–1558 (2013).
Nik-Zainal, S. et al. Cell 149, 994–1007 (2012).
Bivona, T. G. & Doebele, R. C. Nature Med. 22, 472–478 (2016).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Cell 168, 707–723 (2017).
Gao, D. et al. Cell 159, 176–187 (2014).
van de Wetering, M. et al. Cell 161, 933–945 (2015).
Boj, S. F. et al. Cell 160, 324–338 (2015).
Fujii, M. et al. Cell Stem Cell 18, 827–838 (2016).
Sachs, N. et al. Cell 172, 373–386 (2018).
Sato, T. et al. Gastroenterology 141, 1762–1772 (2011).
 Read the paper: Intra-tumour diversification in colorectal cancer at the single-cell level
Read the paper: Intra-tumour diversification in colorectal cancer at the single-cell level
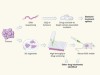 A precision approach to tumour treatment
A precision approach to tumour treatment
 Division hierarchy leads to cell heterogeneity
Division hierarchy leads to cell heterogeneity