- NEWS AND VIEWS
An ode to gene edits that prevent deafness
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 553, 162-163 (2018)
doi: https://doi.org/10.1038/d41586-017-08645-z
References
Gao, X. et al. Nature 553, 217–221 (2017).
Fettiplace, R. & Kim, K. X. Physiol. Rev. 94, 951–986 (2014).
Kawashima, Y. et al. J. Clin. Invest. 121, 4796–4809 (2011).
Kurima, K. et al. Nature Genet. 30, 277–284 (2002).
Zhao, Y. et al. PLoS ONE 9, e97064 (2014).
Vreugde, S. et al. Nature Genet. 30, 257–258 (2002).
Pan, B. et al. Neuron 79, 504–515 (2013).
Carroll, D. Annu. Rev. Biochem. 83, 409–439 (2014).
Jiang, F. & Doudna, J. A. Annu. Rev. Biophys. 46, 505–529 (2017).
Li, H. et al. Nature 475, 217–221 (2011).
Ran, F. A. et al. Nature 520, 186–191 (2015).
Zuris, J. A. et al. Nature Biotechnol. 33, 73–80 (2014).
Tebas, P. et al. N. Engl. J. Med. 370, 901–910 (2014).
Qasim, W. et al. Sci. Transl. Med. 9, eaaj2013 (2017).
Sharma, R. et al. Blood 126, 1777–1784 (2015).
Russell, S. et al. Lancet 390, 849–860 (2017).
 Read the paper: Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents
Read the paper: Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents
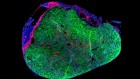
 Gene editing staves off deafness in mice
Gene editing staves off deafness in mice
 Human genes lost and their functions found
Human genes lost and their functions found
 At the heart of gene edits in human embryos
At the heart of gene edits in human embryos