
Abstract
Cells communicate with each other through secreting and releasing proteins and vesicles. Many cells can migrate. In this study, we report the discovery of migracytosis, a cell migration-dependent mechanism for releasing cellular contents, and migrasomes, the vesicular structures that mediate migracytosis. As migrating cells move, they leave long tubular strands, called retraction fibers, behind them. Large vesicles, which contain numerous smaller vesicles, grow on the tips and intersections of retraction fibers. These fibers, which connect the vesicles with the main cell body, eventually break, and the vesicles are released into the extracellular space or directly taken up by surrounding cells. Since the formation of these vesicles is migration-dependent, we named them “migrasomes”. We also found that cytosolic contents can be transported into migrasomes and released from the cell through migrasomes. We named this migration-dependent release mechanism “migracytosis”.
Similar content being viewed by others
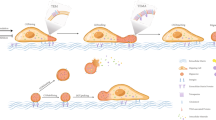


Introduction
Cells release cellular contents through various mechanisms. The contents of secretory vesicles are released from cells by exocytosis when vesicles fuse with the plasma membrane1,2, and exosomes, a group of cell-derived vesicles present in many types of biological fluids, are released from cells by fusion of multivesicular bodies (MVBs) with the plasma membrane3,4,5.
Long projections from the surface of cells were first described in 1945 by a classic study using transmission electron microscopy (TEM)6. In 1963, Taylor and Robbins7 conducted a detailed light microscopy and TEM study in which they documented the appearance of long tubular structures as migrating cells retracted from the substratum. They named these structures “retraction fibrils”7, which were renamed as “retraction fibers” later. To this day, retraction fibers have received little attention despite their widespread presence in different cell types.
In this study, we report the discovery of a cell migration-dependent mechanism for releasing cellular contents, which we named “migracytosis”. We found that during migration, a cell will leave retraction fibers behind it, and vesicles with diameters up to 3 μm grow on the tips or at the intersections of retraction fibers. These vesicles, which we named “migrasomes”, contain numerous smaller vesicles, with diameters of about 50-100 nm. During migrasome biogenesis, there is an initial phase of rapid growth, followed by a relatively stable period. Eventually, retraction fibers break and migrasomes are released into the medium or directly taken up by surrounding cells. Migracytosis is cell migration-dependent, and formation of migrasomes is dependent on both migration and actin polymerization. Migracytosis and migrasomes are present in many cell types, and can occur in cells grown in a 3D matrix or on top of other cells. TEM analysis reveals that migrasome-like structures are present in various tissues. Cytosolic contents are actively transported into migrasomes and subsequently released from cells. Thus, migracytosis is a new cellular process and migrasomes are previously undiscovered organelles which may play important roles in cell-cell communication.
Results
Identification of an extracellular membrane-bound vesicular structure
Using TEM, we observed membrane-bound vesicular structures in the extracellular space around normal rat kidney (NRK) cells (Figure 1A). The distance of these structures from the cell periphery varies considerably. They tend to cluster on one side of the cell, and TEM observations of the bottom of cells revealed that the structures are connected to or closely associated with retraction fibers (Figure 1B). The majority of these membrane structures are oval shaped, with diameters from 0.5 μm to 3 μm, and they contain numerous smaller vesicles. The number of smaller vesicles varies greatly; some structures contain up to 300 vesicles, while most contain fewer than 10 (Figure 1C and 1D). Overall, these structures resemble opened pomegranates. To study how these pomegranate-like structures (PLSs) relate to cells, we carried out a scanning electron microscopy (SEM) study and observed membrane-bound structures with similar shape, size, and position relative to the cell as the PLSs we identified by TEM. SEM revealed that all of these structures are attached to retraction fibers (Figure 1E).

The structure of PLSs revealed by TEM and SEM. (A) TEM image of an ultra-thin section of a NRK cell cultured on a 35 mm dish for 12 h. PLSs are indicated by arrows. Scale bar, 5 μm. Enlarged PLSs are shown in the right panels. Scale bar, 200 nm. (B) TEM image of the lowest ultra-thin section (in contact with the culture dish) of a NRK cell. Scale bar, 5 μm. Enlarged PLSs are shown in the right panels. Scale bar, 200 nm. (C) TEM images of individual PLSs containing different numbers of vesicles. Scale bar, 200 nm. (D) PLSs from A were quantified for their size and the number of small internal vesicles. 120 PLSs were examined. (E) NRK cells were grown on cover slips and observed by field emission scanning electron microscopy. Boxed regions in the top left panel are enlarged in the small panels. Scale bar, 1 μm. (F) TEM images of PLSs in NRK cells and in the isolated PLS fraction. Scale bar, 500 nm.
Purification of PLSs
Since we could not find any reference to these PLSs in the literature, we decided to characterize them in depth. First, we needed a marker which would allow us to carry out live-cell imaging to monitor the behavior of the PLSs. To achieve this goal, we isolated PLSs through a fractionation procedure based on density-gradient centrifugation. TEM confirmed that structures with the same morphological characteristics of PLSs were highly enriched in one of the fractions (Figure 1F).
Identification of markers for PLSs
Our strategy to identify PLS markers involved identifying PLS proteins by mass spectrometry (MS) analysis, then tagging these proteins with GFP and monitoring the fluorescently-labeled proteins by live-cell imaging (Figure 2A). We carried out MS analysis on the enriched PLS fraction. The majority of proteins identified in PLSs are membrane-localized proteins, but cytoskeleton-related proteins are also common (Figure 2B).

Identification of PLS markers. (A) Outline of the screening strategy. MS, mass spectrometry. (B) Classification of the proteins identified by MS according to their function (left) and subcellular localization (right). (C) TSPAN4-mCherry-expressing NRK cells were cultured and then observed by confocal microscope. Scale bar, 5 μm. Enlarged PLSs are shown in the right panels. (D) Statistics of percentage of cells containing PLS and PLS number per cell in PLS-containing cells. TSPAN4-GFP-expressing NRK cells were cultured for 12 h and observed by confocal microscope. The upper panel show the percentage of cells containing PLS in total cells. The lower panel show the PLS number per cell. n = 150 from three independent experiments. (E) Correlative confocal and TEM images of the same NRK cell expressing TSPAN4-mCherry. TSPAN4-mCherry-expressing NRK cells were cultured for 12 h and observed by confocal microscope, then fixed in 2.5% glutaraldehyde for 10 min before preparing ultra-thin sections for observation by TEM. The TEM micrograph (right panel) shows the white boxed region in the confocal image (left panel). An enlarged PLS is shown in the top right of the TEM image. Scale bar, 5 μm.
To screen for PLS markers, we chose a variety of proteins from those identified by MS, and tagged them using GFP. When we checked the localization of these proteins by confocal microscope, we identified Tetraspanin-4 (TSPAN4) as the clearest PLS marker. We established NRK cell lines in which TSPAN4-GFP or TSPAN4-mCherry are stably expressed.
In cells reaching confluence, TSPAN4 mainly has a diffused cellular localization pattern with some of the protein enriched on the plasma membrane and intracellular vesicles (Supplementary information, Figure S1). However, when cultured in low-confluence conditions, the cells have enough space to form long, elaborate retraction fibers. TSPAN4 is enriched on these fibers and is highly enriched on ring-like structures near the fiber ends (Figure 2C). To validate that the TSPAN4-positive ring-like structures in the confocal images are the PLSs in the TEM images, we carried out correlative confocal and TEM analysis (Figure 2E). We found that the TSPAN4-positive ring-like structures are indeed the PLSs observed by TEM (Figure 2D).
The life cycle of PLS
3D reconstruction revealed that each PLS has a bubble-like structure with its bottom attached to a retraction fiber (Figure 3A). Next, we studied the biogenesis of PLSs by time-lapse imaging of stable TSPAN4-GFP-expressing cell lines. We cultured the cells at low confluence, and collected time-lapse images. We found that PLSs formed at fixed sites on retraction fibers and showed a characteristic growth pattern. After an initial phase of rapid growth, the PLSs reached their maximum size, which occurred while the retraction fibers themselves were relatively stable, then entered a relatively stable period without changing size (Figure 3B and 3C, and Supplementary information, Movie S1). Eventually, the retraction fibers broke up, and the PLSs were released into the medium where they eventually disappeared (Figure 3D and Supplementary information, Movie S2). The average lifespan of a PLS is about 400 min (Figure 3E).

Formation and release of PLSs. (A) 3D reconstruction of PLSs. TSPAN4-GFP-expressing NRK cells were cultured, then observed using a DeltaVision microscope. 3D reconstructions were generated by IMARIS software. Yellow, retraction fibers; red, PLSs. The right panels show enlarged regions of interest from the left panel. T, top view; L, lateral view; B, bottom view. Scale bar, 5 μm. (B) Formation of PLSs. TSPAN4-GFP-expressing NRK cells were cultured for 12 h, and time-lapse images were acquired with a NIKON A1 confocal microscope. The right panels show time-lapse images of three PLSs. The left panel shows a wide-field view at 120 min. Images were captured every 10 min for 12 h. Scale bar, 5 μm. (C) Individual PLSs from B were quantified for change of diameter over time. (D) The disintegration of retraction fibers and the release of PLSs. TSPAN4-GFP-expressing NRK cells was cultured for 12 h, and time-lapse images were acquired with a NIKON A1 confocal microscope. The main panel shows a PLS on a retraction fiber behind a migrating cell. Images were captured every 10 min for 12 h. Scale bar, 5 μm. Enlarged images of the same PLS at different times are shown in the right panels. (E) Duration of PLSs. Time-lapse images of TSPAN4-GFP-expressing NRK cells were acquired as described in D and the lifetime of individual PLSs was quantified. Error bar, SEM.
Biogenesis of PLSs is migration-dependent
Time-lapse imaging revealed that the formation of PLSs is likely related to cell migration. When a cell migrates forward, it leaves a network of retraction fibers behind it, and about 40 min after the cell moves away from its original position, PLSs start to appear on the tips or intersections of the retraction fibers (Figure 4A and Supplementary information, Movie S3). When a cell migrates further from its original position, the retraction fibers keep elongating until they break, and the PLSs are then visible for an additional period of time before disappearing (Supplementary information, Movie S3). Since PLSs grow on retraction fibers, and retraction fibers are localized along the migration path of cells, we speculated that formation of PLSs is migration-dependent.

Formation of PLSs is dependent on cell migration. (A) TSPAN4-GFP-expressing NRK cells were cultured for 12 h and time-lapse images were acquired at the indicated times with a NIKON A1 confocal microscope. Images were captured every 10 min for 12 h. Scale bar, 10 μm. (B) Fibronectin (FN) increases the formation of PLSs. TSPAN4-GFP-expressing NRK cells were cultured for 12 h on 35 mm glass-bottom dishes pretreated with 10 μM FN or on untreated dishes (control). Cells were observed by confocal microscope. Scale bar, 10 μm. (C) Cells from B were assessed for the number of PLSs per cell. Y axis, number of PLSs in treated cell/number of PLSs in control cell. n = 100 cells from three independent experiments. Error bars indicate the SD. (D) TSPAN4-GFP-expressing NRK stable cell lines were transfected with NC (control) or SHARPIN siRNA. Cells were cultured for 12 h and observed by time-lapse confocal microscope. Images were captured every 3 min for 4 h. Scale bar, 10 μm. (E) Cells from D were assessed for the number of PLSs. Y axis, number of PLSs in SHARPIN-knockdown cell/number of PLSs in control cell. n = 100 cells from three independent experiments. Error bars indicate the SD. (F) TSPAN4-GFP-expressing NRK cell were treated with or without Blebbistatin and Dynasore and observed by time-lapse confocal microscope. Images were captured every 3 min for 4 h. The formation of individual PLSs within 5 regions of interest (indicated by colored boxes in the left panels) is shown in the small panels. Each region was monitored for 240 min. Scale bar, 10 μm. (G) Cells from F were assessed in a blind fashion for the total number of PLSs per cell (upper panel) and the number of newly-formed PLSs per cell (lower panel) during the observation period and quantified. Y axis, number of PLSs (or newly-formed PLS) in treated cell/number of PLSs (or newly-formed PLSs) in control cell. C, untreated (control) cells; BS, + Blebbistatin; DS, + Dynasore. n = 30 from three independent experiments. Error bars indicate SD.
To test this hypothesis, we coated culture dishes with fibronectin, which is known to increase the migration of cells. We found that cells grown on fibronectin-coated culture dishes indeed move faster and have a markedly increased number of PLSs (Figure 4B and 4C, and Supplementary information, Figure S2). Increasing cell migration by knocking down SHARPIN, a known negative regulator of integrin-dependent cell migration8, also increased the migration speed and the number of PLSs (Figure 4D and 4E, and Supplementary information, Figure S2).
To further validate the hypothesis, we treated cells with the dynamin inhibitor 'Dynasore' and the myosin II inhibitor 'Blebbistatin', both of which have been widely used to block cell migration9,10. We found that these migration inhibitors markedly reduced the number of PLSs, and close analysis revealed that the formation of new PLSs was largely blocked (Figure 4F and 4G, and Supplementary information, Movies S3-S5). Thus, the formation of PLSs is dependent on cell migration. For this reason, we named them “migrasomes”.
Formation of migrasomes is dependent on polymerization of actin
TEM analysis revealed fibrous bundles on the section of the retraction fiber which is closest to the migrasome. On the bottom of the migrasome, the bundle is looser, and individual fibers, similar to polymerized actin fibers, are clearly visible (Figure 5A). To test whether these fibrous structures are indeed actin, we generated a NRK cell line in which TSPAN4-mCherry and LifeAct-GFP, a marker for polymerized actin, are stably expressed. We found that LifeAct is present in some migrasomes (Figure 5B). In many cases, LifeAct is closely associated with the membrane of migrasomes (Figure 5B).

Actin polymerization is required for migrasome formation. (A) TEM images of individual PLSs. Scale bar, 500 nm. (B) NRK cells stably expressing TSPAN4-mCherry were transfected with LifeAct-EGFP, then cultured for 12 h and observed by confocal microscope. (C) NRK cells stably expressing TSPAN4-mCherry were cultured for 12 h with or without Cytochalasin B, CK636 and Latruculin A, and observed by time-lapse confocal microscope. The formation of individual PLSs in 5 regions of interest (indicated by colored boxes in the left panels) is shown in the small panels. Each region was monitored for 550 min. Time interval, 6 min 20 s. Scale bar, 10 μm. (D) Cells from C were assessed in a blind fashion for the total number of migrasomes per cell (left panel) and the number of newly-formed migrasomes per cell (right panel) during the observation period and quantified. Y axis, number of migrasomes in treated cell/number of migrasomes in control cell. Cyto B, Cytochalasin B; Lat A, Latruculin A. n = 30 from three independent experiments. Error bars indicate SD.
To directly test the role of actin in migrasome formation, we treated cells with Cytochalasin B and Latrunculin A, two inhibitors of actin polymerization, and CK636, an inhibitor of the Arp2/3 complex, which blocks formation of branched actin networks. We found that these inhibitors reduce the number of migrasomes by preventing the formation of new migrasomes (Figure 5C and 5D, and Supplementary information, Movies S6-S8). We are aware that the role of actin in migrasome formation may be complex, as some of these actin polymerization inhibitors can also inhibit migration. Nevertheless, these data imply that actin polymerization is likely required for formation of migrasomes, either by affecting cell migration, or by direct involvement in migrasome biogenesis, which is more likely.
Migrasomes are present in multiple cell types
Having established the existence of migrasomes in NRK cells, we next tested whether migrasomes are present in other mammalian cell types. For this purpose, we generated human and mouse TSPAN4-GFP-expressing cell lines. We found that the majority of cells tested can form migrasomes, although the frequency varies (Figure 6A). For cells such as macrophages, primary neuron and embryonic stem cells, which are very hard to monitor using live cell imaging, we carried out TEM analysis, and found that they do have migrasome-like structures (Figure 6B).

Formation of migrasomes in various cell lines and under different growth conditions. (A) MEF (mouse embryonic fibroblast), NIH3T3 (mouse embryonic fibroblast), HaCaT (human keratinocyte), MDA-MB-231 (human breast cancer), HCT116 (human colon cancer), SW480 (human adenocarcinoma), MGC803 (human gastric carcinoma), SKOV-3 (human ovarian adenocarcinoma), and B16 (mouse melanoma) cells were transfected with mouse or human TSPAN4-GFP, cultured for 12 h and observed by confocal microscope. Insets show enlarged regions of interest. Scale bar, 2 μm. (B) Mouse embryonic stem cells, mouse hippocampal neurons and mouse bone marrow-derived macrophages were observed by TEM. Scale bar, 2 μm. (C) TSPAN4-GFP-expressing NRK cells were cultured in a 3D matrix of PEG (polyethylene glycol) mixed with collagen (1 mg/ml) and FN (10 μM/ml) for 20 h, then observed with a DeltaVision microscope. The inset shows an enlarged view of a migrasome. Scale bar, 5 μm. (D) Cells from C were observed by SEM. Scale bar, 5 μm. (E) The gastric carcinoma cell line MGC803 was cultured on glass-bottom dishes to 100% density and stained with CellTracker Green. NRK cells stably expressing TSPAN4-GFP were then added and co-cultured overnight. (F) Cells from E were observed by live-cell imaging (left) and SEM (right). Live-cell imaging was performed with a DeltaVision microscope and 3D reconstructions were generated by IMARIS software. Scale bar, 5 μm. (G) Ultra-thin sections of mouse or rat tissues were observed by TEM. Scale bar, 1 μm. LB, lamellar body; A, Alveolus; En, endothelial cell; C, collagen.
Migrasomes can form on a 3D matrix and on top of cell layers
One concern about migrasomes is that they may be artifacts arising from the tissue culture conditions. To rule out this possibility, we cultured cells on a 3D matrix of polyethylene glycol (PEG) mixed with collagen and fibronectin, which mimics the rigidity of the substratum in vivo. We found that migrasomes still form on this matrix, and thus migrasomes are not an artifact caused by culturing cells on a highly rigid surface such as glass or plastic (Figure 6C and 6D). Next, we tested whether migrasomes can form on top of other cells. We cultured human gastric cancer MGC803 cells until they reached confluence, then NRK cells were added on top. SEM and 3D imaging showed that the NRK cells still formed migrasomes (Figure 6E and 6F). Thus, migrasomes are unlikely to be an artifact arising from the culture conditions.
Observation of migrasome-like structures in vivo
To check whether migrasomes are also present in vivo, we examined various mouse and rat tissues using TEM (Figure 6G). We found migrasome-like structures in various organs including eye, lung and intestine, and these structures tend to be present inside cavity such as blood vessel or pulmonary alveoli. For example, in intestine, migrasome-like structures are inside blood capillaries or lymph capillaries, in the lamina propria of ileum crypts, or in connective tissue. In lung, migrasome-like structures are located inside pulmonary alveoli.
Transport of cellular contents into migrasomes during migrasome formation
As migrasomes form, they grow in size, which prompted us to ask whether cellular contents such as cytosol can enter migrasomes. We overexpressed GFP as a tracer for cytosolic contents in TSPAN4-mCherry-expressing NRK cells, and monitored the formation of migrasomes by time-lapse imaging. Over time, we observed cells moving away from existing migrasomes and new migrasomes forming behind the cells along the migration path. To our astonishment, we found that the GFP signal inside migrasomes kept increasing, which means that cytosol is actively translocated into migrasomes from the main body of the cell, even though the cell is actually moving away from the migrasomes (Figure 7A and 7B, and Supplementary information, Movies S9-S11). As we mentioned above, the migrasome is eventually released into the extracellular environment; thus, the fact that cytosolic contents can actively translocate into migrasomes means that cells can release cytosolic material into the extracellular space through the migrasomes. This represents a mechanism for releasing cellular contents. We named this process “migracytosis”.

Release of cytosolic contents by migrasomes. (A) NRK cells stably expressing TSPAN4-mCherry were transfected with GFP and cultured for 20 h, then time-lapse images were acquired with a NIKON A1 confocal microscope. Images were captured every 6 min 20 s for 4 h. Scale bar, 20 μm. (B) Migrasomes from A were measured for GFP fluorescence intensity using NIKON A1 analysis 4.0 software. (C) Time-lapse images showing NRK-LAMP1-mCherry cells taking up migrasomes from NRK-TSPAN4-GFP cells. LAMP1-mCherry-expressing NRK cells were mixed with TSPAN4-GFP-expressing cells and co-cultured for 12 h, then time-lapse images were acquired with a NIKON A1 confocal microscope. Images were captured every 5 min for 10 h. Insets show enlarged regions of interest. Scale bar, 10 μm.
Migrasomes can be taken up by surrounding cells
During our study, we frequently observed that migrasomes left by one cell can be taken up by another cell. To monitor this uptake process, we mixed TSPAN4-GFP-expressing NRK cells and LAMP1-mCherry-expressing NRK cells. As one cell migrates away from its original position, it leaves migrasomes behind it, then the second cell moves to the location of the migrasomes and takes them up (Figure 7C and Supplementary information, Movie S12).
Discussion
In this study, we report the discovery of a new cellular process, which we named migracytosis. During migracytosis, the migrating cell leaves retraction fibers behind it and migrasomes grow on the tips or intersections of the retraction fibers. Migrasomes contain numerous smaller vesicles and cytosolic contents, and during the growth of migrasomes, translocation of cellular contents into migrasomes occurs. Eventually, the retraction fibers break up and migrasomes are released into the medium or directly taken up by surrounding cells. To the best of our knowledge, our study is the first report of migracytosis and migrasomes.
Both migrasomes and MVBs are large vesicles that contain smaller vesicles in their lumen, and the tetraspanins that mark migrasomes are also abundant on the intraluminal vesicles of MVBs. These observations raise the interesting hypothesis that migrasomes arise from the transport of MVBs into retraction fibers. This hypothesis yields two predictions: (1) migrasomes will have two layers of membrane, with an outer membrane derived from retraction fibers and an inner membrane derived from MVBs; and (2) migrasomes should contain MVB surface markers such as LAMP1. However, the TEM analysis clearly shows that migrasomes only have one membrane (Figure 1A). As for the LAMP1, although a small amount of LAMP1 can be observed on migrasome in cells expressing an extremely high level of LAMP1, for the vast majority of LAMP1-expressing cells, there is no LAMP1 on migrasome (Figure 8A). Thus, it is unlikely that migrasomes arise from intact MVBs that are transported into retraction fibers. It still remains to be determined whether the intraluminal vesicles of MVBs participate in migrasome biogenesis, although in that case, the intraluminal vesicles would have to find a way to escape the MVB to overcome the topological barrier (Figure 8B).

Migrasomes do not contain the MVB surface marker LAMP1. (A) NRK cells stably expressing TSPAN4-GFP were transfected with LAMP1-mCherry, then cultured for 12 h and observed by confocal microscope. Insets show migrasomes. (B) Illustration of the relationship between exosomes, MVBs and migrasomes.
At first glance, migracytosis may have some similarity with the release of exosomes. However, a close look reveals that there are fundamental differences between these two processes. Exosomes are released from cells by fusion of MVBs with the plasma membrane. In contrast, migracytosis involves translocation of cytoplasmic material into migrasomes, then release of the migrasomes by breaking the retraction fibers. Thus, migracytosis and exosome release are mechanistically different processes. By definition, exosomes are cell-derived vesicles present in biological fluids, and most analyses on exosomes are carried out by purifying exosomes from biological fluids or culture medium without investigating where the vesicles are generated. Thus, it is possible that at least some of the exosomes reported previously in the literature may actually be released by migrasomes.
Migrating Dictyostelium discoideum cells have been shown to leave behind adenylyl cyclase (ACA)-containing vesicles11. The authors proposed that these vesicles are exosomes, formed by fusion of external membranes of the MVBs with the plasma membrane, and that they are likely involved in formation of the head-to-tail array of migrating Dictyostelium discoideum cells. This study does not show whether the ACA-containing vesicles are connected to retraction fibers, whether these vesicles are directly shedding from cell as authors proposed or grown on retraction fibers in a manner similar to migrasome, and whether cytosolic contents can continue to be transported into developing vesicles even after cells move away from these vesicles, which is the key definitive feature of migrasomes and the concept of migracytosis. The authors did not examine how these vesicles were formed and thus it remains possible that they are related to migrasomes. To test this, it will be important to determine whether they grow from retraction fibers and whether cytosolic content is transported into them along these fibers, which are key defining features of migrasomes and the concept of migracytosis. Clearly, in light of our discovery, it will be interesting to determine how these vesicles are related to migrasomes.
What is the function of migracytosis and migrasomes? So far, we know that cellular contents, including vesicles and cytosolic factors, can be released into the extracellular space through migracytosis. Thus, the primary function of migracytosis is probably cell-cell communication, similar to mechanisms such as exocytosis and exosome release. The unique way that migracytosis releases cellular contents at a specific location, so that they can be taken up by other cells which travel to that site, means that spatial and biochemical information from outgoing cells can be acquired by incoming cells. Given the fact that many important physiological functions, such as the formation of neuronal networks and innate and adaptive immune responses, require localized communication between cells, we will not be surprised if future work reveals that migracytosis plays an important role in these processes.
Migrasome is a membrane-bound cellular structure with characteristic morphological features, and cells can use migrasomes to release cellular contents including vesicles and cytosolic proteins. In cell biology, an organelle is a specialized subunit of a cell which carries out a specific function. Another key feature of an organelle is that it is usually enclosed within its own membrane. Thus, according to these criteria, we would like to propose that migrasome is a new organelle.
Materials and Methods
Mass spectrometric analysis
Protein bands on SDS-PAGE gels were de-stained and in-gel digested with sequencing grade trypsin (10 ng/μL trypsin, 50 mM ammonium bicarbonate, pH 8.0) overnight at 37 °C. Peptides were extracted with 5% formic acid/50% acetonitrile and 0.1% formic acid/75% acetonitrile sequentially and then concentrated to ∼20 μl. The extracted peptides were separated by an analytical capillary column (50 μm × 10 cm) packed with 5 μm spherical C18 reversed phase material (YMC, Kyoyo, Japan). An Agilent 1100 series binary pump system (Agilent Technologies, Santa Clara, CA, USA) was used to generate the following HPLC gradient: 0%-5% B in 5 min, 5%-40% B in 70 min, 40%-100% B in 10 min (A = 0.2 M acetic acid in water, B = 0.2 M acetic acid/70% acetonitrile). The eluted peptides were sprayed into a LTQ mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) equipped with a nano-ESI ion source. The mass spectrometer was operated in data-dependent mode with one MS scan followed by five MS/MS scans for each cycle. Database searches were performed on an in-house Mascot server (Matrix Science, London, UK) against IPI (International Protein Index) rat protein database. Methionine oxidation was set as variable modification.
Reagents
Fibronectin (PHE0023) was purchased from Gibco. Blebbistatin (D00155970) and Dynamin Inhibitor Dynasore (D00098387) were purchased from Calbiochem. CytoB (C6762), CK636 (C7374), and Latruculin A (L5163) were from Sigma-Aldrich.
Cell culture and transfection
NRK cells were cultured in DMEM (Life Technologies) supplemented with 10% FBS (5% CO2). Cells were transfected with 200 pmol RNAi or a total of 2 μg DNA via Amaxa nucleofection using solution T and program X-001. MGC803, SKOV-3, SW480, MDA-MB-231, HCT116, and B16 were kind gifts from Dr Jijie Chang. NIH3T3 was from Dr Peng Li, and HaCaT was from Dr Yeguang Chen.
To generate the NRK-TSPAN4-GFP stable cell line, NRK cells were transfected with lentivirus and then the cells were selected in medium containing 10 μg/ml Blasticidin. To generate the NRK-TSPAN4-mCherry stable cell line, NRK cells were transfected with 2 μg TSPAN4-mCherry vector and then selected with 1 mg/ml G418.
Constructs
TSPAN4-GFP, Stx4-GFP, Stx7-GFP, Cotl1-GFP, VPS28 homolog-GFP, Vapb-GFP, Vamp2-GFP, Vti1a-GFP, Tmem93-GFP, Tmem85-GFP, Syngr2-GFP, Rab5c-GFP were cloned into the HindIII/AgeI sites of the pEGFP-N1 vector. Mouse and human TSPAN4-GFP, and Rat, mouse and human TSPAN4-mCherry were also cloned into the HindIII/AgeI sites of the pmCherryRed-N1 vector. Rat, mouse and human TSPAN4-GFP and TSPAN4-mCherry were constructed using ViraPower Promoterless Lentiviral Gateway Kits (K5910-00, Invitrogen).
Preparation of small interfering RNA (siRNA)
SHARPIN (Rat) RNAis were synthesized by GenePharma (China). The Sharpin sequence was: 5′-GATAGGAAATTAGGATGCTTGTTTC-3′. The primer sequences for Q-PCR were as follows: forward, 5′-CCGAGAAGTTTCAGGACACA-3′, and reverse, 5′-GGAAGTTGCAGTAAGGGGAT-3′.
Lentivirus transfection
Plasmid construction and lentivirus transfection were performed using ViraPower Promoterless Lentiviral Gateway Kits (Invitrogen) according to the manufacturer's instructions.
Electron microscopy
Field emission scanning electron microscopy For the conventional fixation procedure, the culture cells were grown on coverslips, fixed with 2.5% glutaraldehyde in PB buffer for 2 h at room temperature, washed three times with PB buffer and then post fixed with 1% osmium containing 1.5% potassium ferrocyanide for 60 min at room temperature. All samples were then dehydrated with a graded series of ethanol (50%, 70%, 90%, 95%, and 100%) for 8 min each. After changing ethanol with tert-Butanol, samples were frozen at −20 °C, then dried with a freeze drier. The dried samples were coated with an approximately 10-nm-thick gold film by sputter coating before examination with a field emission scanning electron microscope using an SE detector at an acceleration voltage of 3 kV.
Transmission electron microscopy For morphological analysis of cultured cells, samples were grown on 35 mm dishes and fixed using a conventional fixation procedure (see above). Samples were then dehydrated with a graded series of ethanol (50%, 70%, 90%, 95%, and 100%) for 8 min each. Samples were infiltrated with and embedded in SPON12 resin. After polymerizing for 48 h at 60 °C, 70-nm-thick ultrathin sections were cut using a diamond knife, and then picked up with Formvar-coated copper grids (100 mesh). The sections were double stained with uranyl acetate and lead citrate. After air drying, samples were examined with a transmission electron microscope H-7650 at an acceleration voltage of 80 kV.
Correlative confocal and electron microscopy Live-cell dishes with photo-etched gridded coverslips were used under a confocal microscope to collect 1-2 bright field and confocal images to document the arrangement of the cells at different magnifications. After that, the cells were fixed immediately following the standard EM protocol. After polymerization, the coverslips were removed from the bottom of the live-cell dishes and then processed for sectioning and staining as detailed above.
Live-cell imaging
The night before imaging, cells were cultured in 35 mm glass-bottom dishes coated with fibronectin (10 μM/ml). Images were acquired using NIKON A1 and Olympus FV-1000 confocal microscopes.
Cell fractionation
Cell fractionation was performed by Iodixanol-sucrose density gradient centrifugation, using Optiprep (Sigma-Aldrich, D1556). Cells were collected and lysed in extraction buffer (Sigma-Aldrich, E1156), then the samples were centrifuged at 1000× g for 10 min and then at 2000× g for 20 min. The supernatant, containing the crude migrasome fraction, was fractionated at 150000× g for 4 h in a multi-step optiprep dilution gradient. The gradient was: 2%, 5%, 8%, 10%, 12%, 15%, sample (19%), 25%, 30%. Fractions are prepared for TEM observation to determine fractions in which PLSs are enriched.
3D cell culture
Cells were suspended in DMEM supplemented with 10% FBS, then collagen and fibronectin were added and mixed with the cell suspension. 15-30 μl cell suspension were dropped onto a PEG 3D matrix on 35 mm glass-bottom dishes. After 30 min of incubation at 37 °C in 5% CO2, 2 ml cell culture medium was added. The cells were cultured for 12 h and imaged using a DeltaVision microscope.
Statistical analysis
Statistical analysis was performed in GraphPad Prism. All data were obtained from independent experiments. Error bars in the figures represent the standard deviation (SD). n values are specified in the figure legends.
References
Schneider FH, Smith AD, Winkler H . Secretion from the adrenal medulla: biochemical evidence for exocytosis. Br J Pharmacol Chemother 1967; 31:94–104.
Nagasawa J, Douglas WW, Schulz RA . Ultrastructural evidence of secretion by exocytosis and of synaptic-vesicle formation in posterior pituitary glands. Nature 1970; 227:407–409.
Trams EG, Lauter CJ, Salem N, Heine U . Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim Biophys Acta 1981; 645:63–70.
Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C . vesicle formation during reticulocyte maturation - association of plasma-membrane activities with released vesicles (exosomes). J Biol Chem 1987; 262:9412–9420.
Kowal J, Tkach M, Théry C . Biogenesis and secretion of exosomes. Curr Opin Cell Biol 2014; 29:116–125.
Porter KR, Claude A, Fullam EF . A study of tissue culture cells by electron microscopy - methods and preliminary observations. J Exp Med 1945; 81:233–246.
Taylor AC, Robbins E . Observations on microextensions from the surface of isolated vertebrate cells. Dev Biol 1963; 6:660–673.
Rantala JK, Pouwels J, Pellinen T, et al. SHARPIN is an endogenous inhibitor of beta1-integrin activation. Mol Biol Cell 2011; 22:1315–1324.
Eleniste PP, Huang S, Wayakanon K, Largura HW, Bruzzaniti A . Osteoblast differentiation and migration are regulated by dynamin GTPase activity. Int J Biochem Cell Biol 2014; 46:9–18.
Duxbury MS, Ashley SW, Whang EE . Inhibition of pancreatic adenocarcinoma cellular invasiveness by blebbistatin: a novel myosin II inhibitor. Biochem Biophys Res Commun 2004; 313:992–997.
Kriebel PW, Barr VA, Rericha EC, Zhang GF, Parent CA . Collective cell migration requires vesicular trafficking for chemoattractant delivery at the trailing edge. J Cell Biol 2008; 183:949–961.
Acknowledgements
We are grateful to Nikon instruments (Shanghai) and Jinyu Wang at the Nikon-Tsinghua bio-imaging Core Facility for providing technical support. This work was supported by the National Natural Science Foundation of China (31125018, 31030043 and 31321003), the National Basic Research Program of China (973 program; 2010CB833704, 2011CB910100), and Tsinghua University Grants (2010THZ0, 2009THZ03071) to Li Yu.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Cell confluence affects the formation of PLS. (PDF 227 kb)
Supplementary information, Figure S2
Validation of cell migration inhibitors. (PDF 15 kb)
Supplementary information, Figure S3
CLEM analysis. (PDF 483 kb)
Supplementary information, Movie S1
Formation of PLSs. TSPAN4-GFP-expressing NRK cells were cultured for 12 h and time-lapse images were acquired using a NIKON A1 confocal microscope. Images were captured every 10min for 12h. Related to Figure 3B. (AVI 5807 kb)
Supplementary information, Movie S2
The disintegration of retraction fibrils and the release of PLSs. TSPAN4-GFP-expressing NRK cells were cultured for 12 h and time-lapse images were acquired using a NIKON A1 confocal microscope. Images were captured every 10min for 12h. Related to Figure 3D. (AVI 14705 kb)
Supplementary information, Movie S3
Formation of PLSs is dependent on cell migration. TSPAN4-GFP-expressing NRK cells were cultured for 12 h and time-lapse images were acquired using a NIKON A1 confocal microscope. Images were captured every 10min for 12h. Related to Figure 4A. (AVI 13972 kb)
Supplementary information, Movie S4
Formation of PLSs is dependent on cell migration. TSPAN4-GFP-expressing NRK cells were treated with or without Blebbistatin and Dynasore and observed by time-lapse confocal microscopy. Movie S4, control cell; S5, Blebbistatin; S6, Dynasore. Images were captured every 3min20s for 4h. Related to Figure 4D. (AVI 31129 kb)
Supplementary information, Movie S5
Formation of PLSs is dependent on cell migration. TSPAN4-GFP-expressing NRK cells were treated with or without Blebbistatin and Dynasore and observed by time-lapse confocal microscopy. Movie S4, control cell; S5, Blebbistatin; S6, Dynasore. Images were captured every 3min20s for 4h. Related to Figure 4D. (AVI 34716 kb)
Supplementary information, Movie S6
Formation of PLSs is dependent on cell migration. TSPAN4-GFP-expressing NRK cells were treated with or without Blebbistatin and Dynasore and observed by time-lapse confocal microscopy. Movie S4, control cell; S5, Blebbistatin; S6, Dynasore. Images were captured every 3min20s for 4h. Related to Figure 4D. (AVI 44332 kb)
Supplementary information, Movie S7
Actin polymerization is required for migrasome formation. NRK cells stably expressing TSPAN4-mCherry were cultured for 12 h and observed by confocal microscopy. Cells were treated with Cytochalasin B, CK636 and Latrunculin A and observed by time-lapse confocal microscopy. Movie S7, control; S8, Cytochalasin B; S9, CK636; S10, Latrunculin A. Images were captured every 6min20s for 9h. Related to Figure 5C. (AVI 5312 kb)
Supplementary information, Movie S8
Actin polymerization is required for migrasome formation. NRK cells stably expressing TSPAN4-mCherry were cultured for 12 h and observed by confocal microscopy. Cells were treated with Cytochalasin B, CK636 and Latrunculin A and observed by time-lapse confocal microscopy. Movie S7, control; S8, Cytochalasin B; S9, CK636; S10, Latrunculin A. Images were captured every 6min20s for 9h. Related to Figure 5C. (AVI 34204 kb)
Supplementary information, Movie S9
Actin polymerization is required for migrasome formation. NRK cells stably expressing TSPAN4-mCherry were cultured for 12 h and observed by confocal microscopy. Cells were treated with Cytochalasin B, CK636 and Latrunculin A and observed by time-lapse confocal microscopy. Movie S7, control; S8, Cytochalasin B; S9, CK636; S10, Latrunculin A. Images were captured every 6min20s for 9h. Related to Figure 5C. (AVI 47974 kb)
Supplementary information, Movie S10
Actin polymerization is required for migrasome formation. NRK cells stably expressing TSPAN4-mCherry were cultured for 12 h and observed by confocal microscopy. Cells were treated with Cytochalasin B, CK636 and Latrunculin A and observed by time-lapse confocal microscopy. Movie S7, control; S8, Cytochalasin B; S9, CK636; S10, Latrunculin A. Images were captured every 6min20s for 9h. Related to Figure 5C. (AVI 5130 kb)
Supplementary information, Movie S11
Release of cytosolic contents by migrasomes. NRK cells stably expressing TSPAN4-mCherry were transfected with GFP and cultured for 20 h Time-lapse images were acquired with a NIKON A1 confocal microscope. Images were captured every 6min20 for 4h. Related to Figure 7A. (AVI 5880 kb)
Supplementary information, Movie S12
NRK-LAMP1-mCherry cells taking up migrasomes from NRK-TSPAN4-GFP cells. LAMP1-mCherry-expressing NRK cells were mixed with TSPAN4-GFP-expressing cells and co-cultured for 12 h. Time-lapse images were acquired with a NIKON A1 confocal microscope. Images were captured every 5min for 10h. Related to Figure 7C. (AVI 21860 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article

Cite this article
Ma, L., Li, Y., Peng, J. et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res 25, 24–38 (2015). https://doi.org/10.1038/cr.2014.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2014.135
