
Abstract
Cytoplasmic dynein 1 is fundamentally important for transporting a variety of essential cargoes along microtubules within eukaryotic cells. However, in mammals, few mutants are available for studying the effects of defects in dynein-controlled processes in the context of the whole organism. Here, we deleted mouse Dlic1 gene encoding DLIC1, a subunit of the dynein complex. Dlic1−/− mice are viable, but display severe photoreceptor degeneration. Ablation of Dlic1 results in ectopic accumulation of outer segment (OS) proteins, and impairs OS growth and ciliogenesis of photoreceptors by interfering with Rab11-vesicle trafficking and blocking efficient OS protein transport from Golgi to the basal body. Our studies show that Dlic1 deficiency partially blocks vesicle export from endoplasmic reticulum (ER), but seems not to affect vesicle transport from the ER to Golgi. Further mechanistic study reveals that lack of Dlic1 destabilizes dynein subunits and alters the normal subcellular distribution of dynein in photoreceptors, probably due to the impaired transport function of dynein. Our results demonstrate that Dlic1 plays important roles in ciliogenesis and protein transport to the OS, and is required for photoreceptor development and survival. The Dlic1−/− mice also provide a new mouse model to study human retinal degeneration.
Similar content being viewed by others


Introduction
Photoreceptor cells in the vertebrate retina are highly polarized light-sensory neurons, which can be divided into several morphologically and functionally distinct compartments: the photoreceptor outer segment (OS) and inner segment (IS) continuing into the perikaryon and the synaptic region. The OS is a specialized primary cilium composed of stacks of disk membranes, which links to the IS by the connecting cilium (CC). The disk membranes bear large quantities of phototransduction proteins and undergo renewal throughout life by the assembly of new disks at the base and by the shedding of older disks from the tip1. All of the OS proteins including rhodopsin are synthesized in the endoplasmic reticulum (ER) located in the IS and perikaryon, and funneled through the Golgi. They are then apically transported to the base of the connecting cilium in a dynein-dependent manner2 and finally to the OS3. Accumulating evidence indicates that mutations that affect OS development and/or protein transport to the OS generally lead to photoreceptor degeneration4.
Cytoplasmic dynein 1 is a major microtubule minus-end-directed motor complex and has been implicated in many important processes, including mitosis, membrane trafficking, organelle positioning, and cell migration within eukaryotic cells5. Dynein 1 consists of four distinct classes of subunits: dynein heavy chain (DHC, DYNC1H1), dynein intermediate chain (DIC, DYNC1I1), dynein light intermediate chain (DLIC, DYNC1LI) and dynein light chain (DLC, DYNL)6. The DHC harbors the ATPase and microtubule motor domains, whereas the remaining subunits play regulatory functions, such as the selection of cargoes. DLIC is highly conserved among different eukaryotic species. In mammals there are two Dlic genes for cytoplasmic dynein 1, Dlic1 (Dync1li1) and Dlic2 (Dync1li2), while in lower eukaryotes there is only one Dlic gene. The function of the DLIC protein was first implicated in the control of the mitotic spindle and the assembly of centrioles by the fact that DLIC1, not DLIC2, specifically interacted with pericentrin (PCNT) in Cos-7 cells7. Mutations in the gene encoding DLIC or depleting its gene product by RNAi were shown to result in a variety of mitotic defects from yeast to mammals8,9,10,11. DLIC1 also participates in intracellular vesicle transport via forming a complex with small GTPases Rab4 and Rab11, respectively12,13. However, the functions of the DLIC subunit in keeping the integrity of dynein and Golgi apparatus and the development of neurons are controversial. It was shown that yeast DLIC and mammalian DLIC1 were not required for the stability of dynein complex9,11,14, but depletion or loss of DLIC in Drosophila cells and Aspergillus nidulans led to destabilization of DHC and DIC10,15. Palmer et al.14 reported that depleting DLIC1 by RNAi in HeLa cells disrupted the Golgi apparatus, but Sivaram et al.11 claimed that it had no effect on the Golgi. Mutating Dlic gene blocks the transport of neuronal retrograde cargoes in worms and flies16,17,18 and results in dendritic and axonal defects in neurons, such as a reduction in the length and number of dendrite branches in Drosophila. However, mice with a point mutation Dlic1N235Y display an increase in dendrite length of cortical neurons (with no changes in branching) and the number of dendrite branches of dorsal root ganglia (DRG) neurons, in addition to its increased anxiety-like behavior and altered gait19. Dlic1N235Y was thought to be a gain-of-function mutation.
To better understand the in vivo function of Dlic1 in mammals, we deleted the Dlic1 gene in mice. We found that Dlic1 is not essential for mouse embryonic development or adult survival. The ablation of Dlic1 in mice results in impaired OS growth and ciliogenesis, and photoreceptor degeneration. Dlic1 deficiency leads to the ectopic accumulation of OS proteins, impaired ER export, disturbance of Rab11-vesicle trafficking, and the reduced protein level and altered distribution of dynein subunits. Our data supports the notion that Dlic1 plays an important role in keeping dynein function and is necessary for photoreceptor development and survival.
Results
Establishment of Dlic1−/− mice
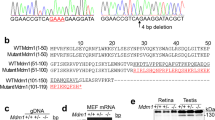
We first generated a modified Dlic1 allele (Figure 1A) by homologous recombination in mouse embryo stem (ES) cells. Targeted ES clones containing the modified allele were identified by PCR and verified by Southern blot (Figure 1B). In the modified Dlic1 allele, a bacterial β-gal reporter gene with a splicing acceptor and a neo expression cassette flanked by FRT sites were inserted after exon 4, and then exon 5 was flanked by loxp sites (Figure 1A). The mosaic mice were created by injecting targeted ES clones into C57BL/6 blastocysts, and then bred with PGK-Cre transgenic mice to generate Dlic1+/galeo mice. In the Dlic1galeo mutant allele, in which the exon 5 was deleted, the expression of β-gal reporter was under the control of endogenous Dlic1 promoter and the β-gal reporter was fused in frame with the first 189 amino acids of DLIC1 protein after splicing. Therefore, the expression pattern of Dlic1 in Dlic1+/galeo mice can be examined using β-gal as a reporter. To establish Dlic1+/− mice, the Dlic1+/galeomice were crossed with Act-Flp-e transgenic mice to remove the β-gal reporter gene and the Neo expression cassette simultaneously (Figure 1A). The Dlic1 gene has 13 exons coding for 523 amino acids. Deletion of exon 5 from Dlic1 resulted in a nonsense mutation at the beginning of exon 6, therefore generating a Dlic1-null mutation. This was further confirmed by analyzing the expression of DLIC1 proteins in the Dlic1−/− mouse brain (Figure 1C).

The generation of Dlic1 knockout mice. (A) Schematic strategy to generate Dlic1−/− mice. WT allele: genomic DNA fragment of the WT Dlic1 gene containing exons 3-6. Targeting vector: schematic structure of the Dlic1 targeting vector. Modified allele: genomic structure of the Dlic1 modified allele after homologous recombination. Dlic1galeo allele: the Dlic1 null allele carrying the β-gal reporter fused in frame to exon 4 of Dlic1 after removing a fragment containing exon 5 by CRE. Dlic1− allele: the Dlic1 null allele derived from the Dlic1galeo allele by removing β-gal and Neo expression cassette by FLP. pgk-DTA and pgk-Neo represent the diphtheria toxin A and the neomycin expression cassettes, respectively. β-gal represents a modified E. coli β-galactosidase gene containing a nuclear localization signal at its N-terminus. The arrows indicate the transcriptional direction of DTA,β-gal and Neo genes. Exons are numbered and depicted by grey boxes. FRT and loxP sequences are represented by white and black arrowheads, respectively. P, N and X indicate the PuvII, NheI and XhoI restriction sites, respectively (Note: Puv II and Nhe I are not unique sites). The gray ellipse under exon 3 in the WT allele depicts the probe used for Southern blot analysis. The expected sizes of the restriction fragments hybridized with the probe are indicated in WT and modified alleles. (B) Southern blot analysis of genomic DNA extracted from WT and targeted ES cell clones and digested with Pvu II and Xho I restriction enzymes. The 3.9 kb and 3.2 kb fragments represent WT and modified alleles, respectively. (C) Western blot analysis of the DLIC1 protein expression in mouse brains using anti-DLIC1antibody. GAPDH was used as loading control. +/+, WT; +/−, Dlic1 heterozygote; −/−, Dlic1 homozygote.
Retinal degeneration in Dlic1−/− mice
Dlic1−/− mice are viable, fertile and show lower body weight (Supplementary information, Figure S1A and S1B). To help to study the physiological role of Dlic1 in mice, we examined the expression pattern of Dlic1 in adult Dlic1galeo/galeo mice using the inserted β-gal as a reporter. We found that Dlic1 was highly expressed in the outer nuclear layer (ONL) of mouse retinas, cerebella and hippocampi (Supplementary information, Figure S1C). The results also revealed that the ONL thickness of Dlic1−/− retinas was reduced compared with the controls (Supplementary information, Figure S1C, top panels), suggesting that the Dlic1 gene may be involved in the development or maintenance of photoreceptor cells.
To further investigate the roles of Dlic1 in the development of mouse retinas, we performed a pathological analysis of Dlic1−/− retinas. As the body weight and the ONL thickness of Dlic1+/− mice are not obviously different from those of wild types, Dlic1+/− mice were used as controls. The results showed that the Dlic1−/− retina displayed a gradual loss of photoreceptor cells. By 12 days after birth (P12), the thickness of the ONL in Dlic1−/− and control retinas was comparable at 11-13 rows of nuclei (Figure 2A and 2B left panel). However, the thickness of ONL of one month (1M) Dlic1−/− retinas was significantly reduced to ∼6 rows of nuclei (Figure 2A and 2B left panel), suggesting a dramatic loss of photoreceptor cells in Dlic1−/− retinas at age 1M. This loss of cells continued as the Dlic1−/− mice aged, but at a much slower rate (Figure 2A and 2B left panel).

Photoreceptor degeneration and impaired OS development in the Dlic1−/− mouse retina. (A) Images of HE-stained cryosections of Dlic1+/− and Dlic1−/− mouse retinas at the indicated ages. (B) Quantitative analysis of the thickness of the ONL (left) and length of the OS (right) in Dlic1+/− and Dlic1−/− mouse retinas at the indicated ages. (C, D) Representative images of TUNEL assay (C) and quantitative analysis (D) of apoptotic cells in the ONL of Dlic1+/− and Dlic1−/− retinas at the indicated ages. (E) Representative fluorescent images of cryosections of P20 Dlic1+/− and Dlic1−/− retinas stained with anti-GFAP show upregulation of GFAP in Dlic1−/− Müller glia. (F) Representative images of PNA-stained cone cells in Dlic1+/− and Dlic1−/− retinas at P12 and P20 show the reduced number and impaired OS development of Dlic1−/− cone cells. Cell nuclei in C, E and F were stained with DAPI. Images are representative retina sections from at least three mice per group. SG = OS + IS; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer; M, month; +/−, Dlic1+/−; −/−, Dlic1−/−. The values in B and D represent the means ± SEM of three mice. ns, not significant; ***P < 0.001. Bar = 50 μm.
Further TUNEL analysis showed that many TUNEL-positive signals were detected in the ONL of P20 (postnatal day 20) Dlic1−/− retinas, while very few to no TUNEL-positive signals were found in the ONL of P12 Dlic1−/− retinas (Figure 2C and 2D). Consistent with the reduced loss of photoreceptor cells in older Dlic1−/− mice, TUNEL signals in the ONL of Dlic1−/− retinas were gradually reduced after one month of age, and only a few TUNEL signals were still detected in 10-month Dlic1−/− retinas (Figure 2C and 2D). These data demonstrate that the gradual loss of the photoreceptors in the Dlic1−/− retina is mainly caused by apoptosis. Furthermore, we also found that the expression of glial fibrillary acidic protein (GFAP) was upregulated in Dlic1−/− retinas (Figure 2E), indicating that Müller cells were activated for proliferation as a result of retinal damage20.
All the results above suggest that ablation of the Dlic1 gene in the mouse results in photoreceptor degeneration.
Ablation of Dlic1 impairs the development of photoreceptor cells
Detailed pathological analysis also revealed that the length of the OS of P12 Dlic1−/− retina was significantly shorter compared with the control although no obvious cell loss in the ONL could be observed in mutant retinas (Figure 2A most left panel, and 2B). The OS length of the mutant retina in aged mice never reached that of their controls (Figure 2A and 2B, right panel). However, ultrastructure analysis with electron microscopy demonstrated that the disk membranes remained well ordered and tightly stacked in the OS of Dlic1−/− photoreceptor cells (Supplementary information, Figure S2). These results suggest that Dlic1 deficiency impairs the OS growth, but has no obvious effect on the disk membrane structures of the OS in the Dlic1−/− retina.
The ONL of mouse retina is composed of ∼95% rod and ∼5% cone nuclei. Thus, the observations of shorter OS length and the reduced ONL cell number in the Dlic1−/− retina strongly suggest that the OS development and survival of rod cells are affected by Dlic1 deficiency. However, it is not clear whether cone cells are also affected in the Dlic1−/− retina. In order to determine the effect of Dlic1 deficiency on cone cells, we performed immunofluorescent staining with FITC-conjugated peanut agglutinin (PNA), which specifically labels cone photoreceptors. Similarly to Dlic1−/− rod cells, the results showed not only a decrease in the OS length of cone cells, but also a dramatic decrease in the number of cone cells in P12 Dlic1−/− retinas (Figure 2F, top panels). The similar phenotypes were also observed in older Dlic1−/− retinas (Figure 2F, bottom panels). However, almost no TUNEL signal could be detected in P12 Dlic1−/− retina (Figure 2C). Therefore, these results suggest that the development of cone cells is also impaired.
Dlic1 deficiency leads to impairment of primary ciliogenesis
The OS of photoreceptor cells is a specialized cilium. Accumulating evidence indicates that mutations in genes involved in ciliogenesis and/or protein transport to the OS generally affect OS development and finally lead to photoreceptor degeneration4. Severe photoreceptor degeneration and shorter OS in Dlic1−/− retinas (Figure 2A-2C) suggest the effects of Dlic1 deficiency on ciliogenesis and/or protein transport to the OS. Therefore, we first verified the effect of Dlic1 deficiency on ciliogenesis by examining the development of connecting cilia in Dlic1−/− photoreceptors at ages of P12 and P20, when photoreceptor OS was growing21. Cryosections of Dlic1−/− and control retinas were immunofluorescent stained with an antibody against acetylated-tubulin (Ac-tubulin), a marker for primary cilia. Notably, we found that the mean cilium lengths were significantly shorter in P12 and P20 Dlic1−/− photoreceptors (Figure 3A-3C, 0.59 ± 0.02 μm (n = 249) and 0.45 ± 0.02 μm (n = 171) for the control and mutant at P12, respectively; 1.01 ± 0.05 μm (n = 117) and 0.84 ± 0.04 μm (n = 136) for the control and mutant at P20, respectively). These results indicate that ciliogenesis is impaired in Dlic1−/− photoreceptor cells.

Dlic1 deletion impairs primary ciliogenesis by disrupting Rab11-vesicle transport. (A, B) Impaired development of the connecting cilium in the Dlic1−/− photoreceptors. Cryosections of the Dlic1+/− and Dlic1−/− retinas at age of P12 (A) and P20 (B) were immunofluorescent stained with anti-Ac-tubulin. (C) Statistical analysis of the length of the connecting cilia in Dlic1+/− and Dlic1−/− mouse photoreceptors at the indicated ages. Length of cilia in five random areas per section were measured and retinas from three mice in each group were used for the analysis. (D, E) Impaired ciliogenesis in Dlic1−/− MEFs. Primary ciliogenesis of Dlic1+/− and Dlic1−/− MEFs was induced by serum starvation. Images of primary cilia stained with anti-Ac-tubulin (D) and statistical analysis of the cilia length (E, left panel) and the percentage of cells with cilia (E, right panel) in serum-starved Dlic1+/− and Dlic1−/− MEF cells (n = 32 and 44 for the control and mutant respectively). (F) Dlic1 deletion does not affect the distribution pattern of PCNT in photoreceptor cells. Cryosections of P5 Dlic1+/− and Dlic1−/− retinas were IF stained with anti-PCNT. (G) IF stained cryosections of P12 Dlic1+/− and Dlic1−/− retinas with anti-Rab11 show the misaccumulation of Rab11 vesicles in the ONL of Dlic1−/− retina. (H) IP-western blot (left pane) and quantitative analyses (right panel) show that loss of Dlic1 leads to a decreased interaction between Rab11 and dynein. Cell nuclei are stained with DAPI. CC, connecting cilium. Images are representative retina sections from at least three mice per group. The values in C, E, H represent the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Bar = 20 μm.
To further ascertain the effect of Dlic1 deficiency on ciliogenesis, we also examined ciliogenesis in Dlic1−/− MEFs. Dlic1−/− and control MEFs were first starved for serum to induce ciliogenesis, and then the number of cells with cilia and the length of primary cilia were evaluated by immunofluorescent staining with anti-Ac-tubulin. The experiments revealed that both the length of primary cilia and the percentage of ciliated cells in Dlic1−/− MEFs were reduced compared with those in the control (Figure 3D and 3E). Therefore, we demonstrate that Dlic1 deficiency impairs primary ciliogenesis in both photoreceptor and MEF cells.
Loss of Dlic1 does not affect apical location of centrioles during ciliogenesis
In mammalian photoreceptor precursor cells, a pair of centrioles complete its migration towards the apical cell membrane by midgestation, and the basal body is developed from the mother centriole and remains in that position22. The apical localization of centrioles is thought to be one of the prerequisites for primary ciliogenesis23,24. As dynein plays a key role in the movement of centrosomes in many cellular events, such as cell division and cell differentiation5, loss of Dlic1 might impair ciliogenesis by affecting the centriole localization. To understand the mechanisms underlying the impairment of ciliogenesis in Dlic1−/− photoreceptor cells, we examined the location of basal bodies in P5 Dlic1−/− retinas by immunofluorescent staining with an antibody against PCNT, which is a component of the basal body complexes of the CC in photoreceptors25. Our study revealed that the basal bodies in both P5 Dlic1−/− and the control retinas were properly localized in the apical region of the IS of photoreceptors (Figure 3F), suggesting that Dlic1 deficiency does not affect the apical localization of basal bodies in photoreceptor cells before the OS starts to develop.
Disrupted Rab11-vesicle transport from the Golgi to the basal body in Dlic1−/− photoreceptors
The Rab11-mediated vesicle transport from the Golgi to the basal body is required for ciliogenesis26,27. Rab11 was linked to cytoplasmic dynein via the direct interaction of Rab11-FIP3 with DLIC1 or DLIC213,28. On the basis of these previous studies, we questioned whether Dlic1 deficiency might impair ciliogenesis by blocking the transport of Rab11-mediated vesicle from the Golgi to cilia due to the reduced interaction between dynein and Rab11. To address the question, we first examined the localization of Rab11 in Dlic1−/− retinas. The results showed that large quantities of Rab11 were localized in the ONL layer (perinuclei) of Dlic1−/− retina, while they were mainly localized in the IS of control retinas (Figure 3G), demonstrating the blockage of Rab11-vesicle transport from the Golgi to cilia. Next we examined whether the interaction between dynein and Rab11 decreased in Dlic1−/− photoreceptor cells by immunoprecipitation and western blot. The analysis showed that the protein level of Rab11 coimmunoprecipitated with dynein was reduced (Figure 3H), indicating that the decreased ability of dynein to capture the Rab11-vesicle was due to Dlic1 deficiency. This result provides at least one explanation for the failure of Rab11-vesicle transport. The results presented above also suggest that Dlic1 deficiency-mediated blockage of Rab11-vesicle transport from the Golgi to cilia is one cause leading to the impairment of ciliogenesis in Dlic1−/− photoreceptor cells.
On the basis of these results, we conclude that Dlic1 deficiency impairs photoreceptor ciliogenesis by blocking the Rab11-vesicle transport to basal bodies, but not by interfering with the apical localization of basal bodies.
Mislocalization of OS proteins in Dlic1−/− photoreceptor cells
The photoreceptor OS contains large amounts of phototransduction proteins. All OS proteins are synthesized in the IS. Impaired transport of OS proteins from the IS to the basal body or insufficient supply of OS proteins will impede OS development, and finally lead to photoreceptor degeneration4,29. In Drosophila, rhodopsin normally colocalizes with Rab11 in vesicles, and the loss of Rab11 activity results in the accumulation of rhodopsin-containing vesicles in the cytoplasm30. We found that Dlic1 deficiency blocks Rab11-vesicle transport from the Golgi to cilia (Figure 3G). Therefore, it is conceivable to postulate that Dlic1 deficiency causes OS underdevelopment by impeding OS protein transport from the IS to the OS of photoreceptor cells. To test our model, we examined the localizations of the OS proteins in Dlic1−/− retinas by immunofluorescent staining. Our results showed that rhodopsin was ectopically accumulated in the cell body especially in the outer plexiform layer (OPL) of P20 Dlic1−/− retina in addition to its normal OS localization (Figure 4A and 4B). We also found that the distribution of another OS protein arrestin displayed a similar ectopic accumulation in P20 Dlic1−/− retinas (Figure 4C). These results demonstrate that Dlic1 deficiency impairs protein transport in photoreceptor cells. We suggest that Dlic1 deficiency-mediated impairment of protein transport provides a mechanism underlying underdevelopment of OS in Dlic1−/− retinas.

Mislocalization of OS proteins in Dlic1−/− photoreceptor cells. (A) Immunofluorescent staining of cryosections of P20 Dlic1+/− and Dlic1−/− retinas with anti-rhodopsin reveals the mislocalization of rhodopsin in Dlic1−/− photoreceptor cells. (B) Higher magnification of P12 mouse retinas immunofluorescent -stained with anti-rhodopsin. Perinuclear mislocalized rhodopsin is only shown in the Dlic1−/− retina. (C) Immunofluorescent-stained cryosections of P20 Dlic1+/− and Dlic1−/− retinas with anti-arrestin. Arrows and arrowheads in A and C indicate the mislocalization of rhodopsin or arrestin in the OPL and the IS, respectively. Cell nuclei were stained with DAPI. Images are representative retina sections from at least three mice per group. Bar = 20 μm.
Dlic1 deficiency does not disrupt the Golgi architecture and may not affect vesicle transport from the ER to Golgi
Rab11 vesicles and rhodopsin were both accumulated in the cytoplasm of Dlic1−/− photoreceptor cells (Figures 3G, 4A and 4B). However, we found that their accumulation patterns in the cytoplasm of Dlic1−/− photoreceptor cells were not exactly the same, e.g., rhodopsin heavily accumulated in the OPL of P20 Dlic1−/− retina, while Rab11 did not. The OS proteins are synthesized in the ER, funneled through the Golgi and then transported to the OS. As Rab11 is implicated in mediating vesicles transport from the Golgi to the basal body26,27, the distinction of accumulation patterns between Rab11 and rhodopsin hints that Dlic1 deficiency may also affect protein transport from the ER to Golgi, in addition to its effect on the protein transport from the Golgi to cilia. Indeed, knocking down Dlic1 but not Dlic2 has been reported to disrupt the Golgi architecture and block ER-to-Golgi transport in HeLa cells14.
On the basis of these previous studies, we assumed that deleting Dlic1 gene also resulted in impairment of protein transport from the ER to Golgi and disruption of the Golgi architecture. To validate the assumption, we first tried to verify the effects of Dlic1 deficiency on the Golgi architecture by immunofluorescent staining with an antibody against giantin, a marker for the Golgi. Our studies showed that the Golgi apparatus was normally concentrated in the proximal portion of the IS in control retinas (Figure 5A, left panel). Our studies also revealed that the distribution of Golgi was not changed and the Golgi architecture appeared intact in Dlic1−/− retinas (Figure 5A, right panel), suggesting that the Golgi apparatus is not disrupted in Dlic1−/− photoreceptor cells. To further confirm the integrity of the Golgi apparatus in Dlic1−/− cells, we performed the same experiment with Dlic1−/− MEFs. Results showed that the Golgi architecture in Dlic1−/− MEFs was not scattered, but assembled normally at the juxtanucleus (Figure 5E, right panel), which was very similar to that in the control (Figure 5E, left panel). These results demonstrate that Dlic1 deficiency does not disrupt the Golgi architecture in either photoreceptor or MEF cells.

The effect of Dlic1 deficiency on the ER-to-Golgi transport in photoreceptors. (A-C) Dlic1 deficiency does not affect the Golgi structure and distribution, but leads to the mislocalization of GalT and calnexin in the OPL. Cryosections of P20 Dlic1+/− and Dlic1−/− retinas were stained with anti-giantin (A), anti-GalT (B) and anti-calnexin (C) separately. Arrows in B and C indicate the mislocalization of GalT and calnexin in the OPL, respectively. (D) Electron micrographs of photoreceptors in P20 Dlic1+/− and Dlic1−/− mice, showing the ER in the IS. Arrows in the left and right panels indicate normal or dilated ER, respectively. (E) Co-immunofluorescent staining of Dlic1+/− and Dlic1−/− MEFs with anti-GalT and anti-Giantin shows the colocalization of GalT and Giantin signals in both mutant and control MEFs. Images are representative retina sections or MEFs from at least three mice per group. Bar = 20 μm (A-C, E); 250 nm (D).
Next, we investigated whether vesicle transport from the ER to Golgi was impaired by examining the localization of 1, 4-galactosyltransferase I (GalT) in Dlic1−/− and control retinas. GalT-containing vesicles cycle slowly between the ER and Golgi31, and GalT localizes at steady state to trans-Golgi cisternae32, thereby, serving as an endogenous indicator of ER-to-Golgi transport. Our immunofluorescent staining with anti-GalT showed that unlike the giantin that was mainly located in the IS of the control retina, GalT was distributed in both the IS and the ONL (perinuclei) of the control retina, indicating that GalT is located at both the ER and Golgi in photoreceptor cells (Figure 5B, left panel). Dlic1−/− retina displayed a GalT distribution pattern very similar to that of the control except for the distribution of GalT extended into the OPL (Figure 5B, right panel). These results indicate that (1) the ER is mislocalized in Dlic1−/− photoreceptor cells, and (2) GalT is not a proper marker for ER-to-Golgi transport in photoreceptor cells.
To evaluate the effect of Dlic1 deficiency on ER-to-Golgi transport, we analyzed the distribution of GalT in MEFs by immunofluorescent staining with anti-GalT. Our results showed that GalT in control MEFs displayed a juxtanuclear pattern strongly reminiscent of the Golgi apparatus, and colocalized with giantin (Figure 5E, left panel). These results are consistent with that previously reported in HeLa or other cells14,32, suggesting that GalT can serve as a marker for ER-to-Golgi transport in MEFs. However, we surprisingly found that GalT in Dlic1−/− MEFs not only displayed a very similar pattern to that of the control, but also colocalized with giantin (Figure 5E, right panel), indicating that Dlic1 deficiency may not affect normal vesicle transport from the ER to Golgi in MEFs, or have very little effect.
Dlic1 deficiency partially blocks ER export
The extension of GalT distribution into the OPL in Dlic1−/− retinas (Figure 5B) strongly indicates a mislocalization of the ER in Dlic1−/− photoreceptor cells. To ascertain the mislocalization of the ER in Dlic1−/− photoreceptors, we examined the ER distribution in Dlic1−/− photoreceptors by immunofluorescent staining with an antibody against calnexin (an ER-specific protein). The results revealed that the ER was accumulated in the OPL of the Dlic1−/− retina in addition to its distribution in the IS and the ONL (perinuclei), while it was only scattered in the IS and the ONL of the control retinas (Figure 5C). Thus, we verified the mislocalization of the ER in Dlic1−/− photoreceptors. We suspect that it is probably due to Dlic1 deficiency-mediated impairment of dynein transport function (see discussion).
As both the ER (Figure 5C) and rhodopsin (Figure 4A) were aberrantly accumulated in the OPL of Dlic1−/− retinas, we suspected that mislocalization of rhodopsin could also be partially because of the aberrant accumulation of rhodopsin in the ER. If this were the case, rhodopsins should be colocalized with the ER marker, calnexin. Indeed, our co-immunofluorescent staining experiments with anti-rhodopsin and anti-calnexin showed that signals of colocalization of these two proteins were detected not only in the OPL, but also in the ONL layer (perinuclei) of Dlic1−/− retinas, while few to none were found in the controls (Supplementary Information, Figure S3). Ultrastructure analysis with electron microscopy also revealed that the ER in Dlic1−/− photoreceptors was dilated (Figure 5D), a cellular phenomenon usually resulting from the aberrant accumulation of unfolded or misfolded proteins in the ER or treatment with an ER stressor33. The results presented above strongly indicate that Dlic1 deficiency partially blocks vesicle export from ER. Therefore, we suggest that the aberrant accumulation of rhodopsin in the ER of Dlic1−/− photoreceptors provides another mechanism underlying mislocalization of rhodopsin in Dlic1−/− retinas.
DLIC1 is essential for the stability and proper distribution of dynein subunits in mouse photoreceptor cells
The mutant phenotypes of defective protein transport from the Golgi to cilium and mislocalization of the ER in the OPL of Dlic1−/− retinas strongly suggest the dysfunction of dynein transport. Loss of the DLIC protein in Drosophila and Aspergillus results in destabilization of both the DHC and DIC and the dynein complex10,15, although depleting mammalian DLIC1 was reported to have no effects on the stability of dynein subunits or integrity of dynein complex11,14. To understand the molecular mechanisms by which the Dlic1 mutation impairs transport function of dynein in photoreceptor cells, we examined the effect of Dlic1 deficiency on dynein in Dlic1−/− retinas. Western blot analysis showed that the protein levels of DHC, DIC and DLC (Tctex-1) were significantly reduced in Dlic1−/− retinas, while the protein levels of DLIC2 was not significantly altered, and dynactin p150 Glued were dramatically increased (Figure 6A). The decrease of DIC protein level was also observed in other tissues/cells of the Dlic1−/− mice, such as brain, liver, and MEFs (Figure 6B and Supplementary information, Figure S4A). To investigate whether Dlic1 deficiency regulates the expression of dynein subunits at transcriptional level, we evaluated the expression of both Dhc and Dic1 at mRNA level by RT-PCR analyses, and found that mRNA expression of Dhc and Dic1 was not significantly altered compared with the control (Supplementary information, Figure S4B). These results suggest that the DLIC1 protein is essential for the stability of dynein DHC, DIC and DLC subunits, and Dlic1 deficiency may impair the transport ability of dynein by destabilizing dynein subunits.

Deletion of Dlic1 disrupts the distribution of dynein in mouse retinas and destabilizes other subunits of dynein. (A) Ablation of Dlic1 leads to the decreased protein levels of dynein subunits in retinal cells. Western blot (left panel) and quantitative (right panel) analyses of the protein levels of dynein subunits in retinal cells. (B) The reduction of DIC level in Dlic1−/− brain, liver and MEFs. Cell lysates from Dlic1+/− and Dlic1−/− retinas (A) or other tissues/cells as shown (B) were immunobloted with the indicated antibodies. GAPDH was used as a loading control. (C, D) Loss of Dlic1 results in the disruption of distribution of DHC and DIC in mouse retinas. Cryosections of P12 Dlic1+/− and Dlic1−/− retinas were immunofluorescent stained with anti-DHC (C) and anti-DIC (D). Cell nuclei were stained with DAPI. Images and blots are representative from at least three mice per group. The values in A represent the means ± SEM of three mice. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001. Bar = 20 μm.
Dynein subunits are apically distributed in the inner segment of photoreceptor cells2,34. The apical distribution of Tctex-1 was demonstrated to be correlated to the apical transport of proteins in polarized MDCK cells35. If the apically distributed dynein is self-driven, then we expect that Dlic1 deficiency should result in the loss or disturbance of the apical distribution of dynein. To address this question, we analyzed the distributions of DHC and DIC in Dlic1−/− retinas by immunofluorescent staining. The results showed that the distributions of DHC and DIC in Dlic1−/− retinas were dramatically disturbed (Figure 6C and 6D). For DHC, the control retina displayed very strong signals in the IS and the OPL; however, the Dlic1−/− retina showed much weaker signals in the IS, although the DHC signal in OPL was similar to that of the control (Figure 6C). While there were strong DIC signals in the IS of the control retina, there were only diminished DIC signals in the IS of the Dlic1−/− retina (Figure 6D). These results strongly demonstrate that retrograde transport of dynein complex without DLIC1 subunit in Dlic1−/− retina is impaired.
Defective ERG responses of the Dlic1−/− photoreceptors
Finally, we determined whether Dlic1 deletion affects retinal function. We used electroretinography (ERG) to analyze the rod- and cone-driven circuits in both scotopic and photopic conditions. Under scotopic and photopic conditions, the ERG detects responses from rod- and cone-driven circuits, respectively. We compared a-wave and b-wave of Dlic1−/− mice under both scotopic and photopic conditions with those of control mice (Figure 7A-7D). Under scotopic conditions, while the average saturated a-wave and b-wave represent 373 μV and 875 μV, respectively, in control mice, the average saturated a-wave and b-wave were reduced to 104 μV and 379 μV correspondingly in Dlic1−/− mice (Figure 7A and 7C). These data illustrate that the rod-driven circuit is significantly affected in the Dlic1−/− mouse retina. Similar to the scotopic ERG responses, photopic ERG responses were weakened both at a-wave and b-wave in Dlic1−/− mice. Under the photopic conditions, the a-wave and b-wave represent 10 μV and 117 μV in control mice and 0.9 μV and 16 μV in Dlic1−/− mice, respectively (Figure 7B and 7D). These results demonstrate that both the scotopic and photopic responses of the Dlic1−/− retina are impaired.

Functional characteristics of Dlic1+/− and Dlic1−/− retinas. (A, B) Saturating ERG responses of the retinas of the mice as indicated to 510 nm flashes at an intensity of -35 log scot. cd. s/m2 under scotopic (A) and photopic (B) conditions. Each trace is the average of individual records from five mice. (C, D) Statistical analyses of the saturating amplitude of a- and b-waves of the mice as indicated under scotopic (C) and photopic (D) conditions. The Dlic1+/− and Dlic1−/− mice were 10-month-old littermates. Values in C and D represent the means ± SEM of five mice. ***P < 0.001.
Discussion
Accumulating evidence demonstrates that mutations in genes encoding motor proteins, such as Kinesin-2 and dynein 2 responsible for anterograde and retrograde intraflagellar transport, respectively, result in ciliopathy36. Tctex-1, a light chain subunit of cytoplasmic dynein 1, has been recently proposed as a component of dynein 2 to negatively regulate cilium length37 and/or participate in cilium disassembly, but not ciliogenesis in a dynein-independent manner38. We have now shown that primary ciliogenesis is impaired in both Dlic1−/− photoreceptor and MEF cells, thus for the first time demonstrating that DLIC1 is a positive regulator of ciliogenesis, and that cytoplasmic dynein 1 also participates in the assembly of primary cilia. We have found that the apical localization of centrioles is not affected before the development of the OS, but Rab11 vesicles were ectopically localized in the ONL of Dlic1−/− retinas. As Rab11-mediated vesicle transport from Golgi to the basal body is required for ciliogenesis, we conclude that Dlic1 deficiency impairs ciliogenesis and OS development by blocking polarized vesicle transport, but not by interfering with the apical localization of basal bodies/centrioles.
Our study shows that Dlic1 deficiency results in ectopic accumulation of Rab11 vesicles in the ONL of retinas. Rab11 vesicle transport from the Golgi to the basal body is dynein dependent13, and Golgi and the basal body are IS features in photoreceptor cells. However, how does Dlic1 deficiency-mediated loss of function for dynein lead to accumulation of Rab11 vesicles in the ONL rather than at an origin or terminus within the IS? In highly polarized photoreceptor cells, microtubules are arranged with their minus ends clustered around a basal body near connecting cilium and their plus ends pointing towards OPL. Many of cargoes or vesicles including endosomes and ER are transported along polarized microtubules bidirectionally with plus- and minus-end motors present on the surface of cargo simultaneously. The direction of the net transport depends on the balance between plus- and minus-end directed motions. Thus, a possible explanation for ectopic accumulation of Rab11 vesicles could be that Dlic1 deficiency results in an imbalance of cargo transportation between plus- and minus-end directions in cells. Impairing or enhancing minus-end motors should improve or reduce motion in the direction of plus-end motors and vice versa39,40. We show here that deletion of Dlic1 results in decreased protein levels of multiple dynein subunits, including DHC, DIC and DLC (Tetcx-1), but not DLC2. Therefore, Dlic1 deficiency reduces the total number of minus-end motors in Dlic1−/− photoreceptor cells, and the plus-end trafficking is then enhanced. This will facilitate the travel of Rab11 vesicles in the plus-end direction and retard their motion in minus-end direction. The final result is that Rab11 vesicles delocalize from their normal IS location and accumulate in the ONL.
Dlic1 deficiency results in ER mislocalizeation in the OPL of mouse retinas. The ER is a highly dynamic organelle that undergoes constant movement and reorganization. Dynein drives the ER mobility towards the minus end of microtubules, while kinesin drives ER movement towards the plus end of microtubules41. Therefore, Dlic1 deficiency-mediated imbalance of cargo transportation between plus- and minus-end directions in cells could also provide an explanation for ER mislocalization in the OPL of Dlic1−/− retinas.
The role of DLIC proteins in the assembly of the core dynein complex has been controversial. The DLIC protein is required for the stability of both the DHC and DIC in Drosophila and Aspergillus, and is able to stabilize the interaction between DHC and DIC in Aspergillus10,15. However, depleting DLIC1 or DLIC211,14 by RNAi in HeLa cells did not affect the protein levels of DHC, DIC and DLC (Tctex-1). We have now found that the deletion of Dlic1 in mouse leads to the destabilization of DHC, DIC and DLC (Tctex-1) in many mouse tissues or cells, such as retinas and MEFs, but does not affect DLIC2. Our findings support the notion that the assembly of core dynein subunits, including the DHC, DIC and DLIC, is an interdependent process10. The discrepancy about the effect of DLIC1 on the stability of the dynein complex or subcomplex between HeLa cells and our Dlic1-deficient mouse cells may result from the different methods used to deplete DLIC1: knockdown by RNAi may not completely eliminate DLIC1 but our Dlic1−/− mouse is a null mutant.
In mammalian cells the formation and budding of COPII-coated vesicles at ER export sites (ERES) is the first step of membrane trafficking in the secretory pathway42. The budded vesicles are then transported from ERES to the ER-Golgi intermediate compartment (ERGIC) in a COPII-dependent step, and at ERGIC they are fused to form vesicular-tubular transport carriers (VTC) and finally transported to the cis-Golgi in a second COPII- and dynein-dependent step43,44. Disruption of interaction between COPII complex and dynactin, a dynein adaptor linking dynein to cargoes, inhibits cargo export from the ER, but does not affect the motility of VTCs44. This study suggests that ER export may be dynein dependent. Here we show mis-accumulation of rhodopsins in dilated ER of Dlic1−/− photoreceptors, suggesting partial blockage of vesicle export from the ER, and providing direct evidence to support the involvement of dynein or dynein subunit in ER export. However, the effect of DLIC on protein transport of dynein from the ER to Golgi is controversial, too. Palmer et al.14 reported that knocking down Dlic1 in HeLa cells resulted in blockage of vesicle export from ER and VTC transport from the ER to Golgi, and the failed VTC transport leads to fragmentation of the Golgi structure; but Sivaram et al.11 showed that depleting DLIC1 in the same cell line had no effect on the Golgi structure at all. Here we demonstrate that the Golgi structure in both Dlic1−/− photoreceptors and MEFs is intact and GalT is steady-stately localized in trans-Golgi cisternae in Dlic1−/− MEFs, strongly indicating that VTC transport is not affected in Dlic1−/− cells. Therefore, we infer that Dlic1 deficiency impairs vesicle export from ER, but not VTC transport from the ER to Golgi. The possible explanations for our results could be (1) the DLIC1 protein may play a specific role in ER export; (2) DLIC1 and DLIC2 proteins are functionally redundant for VTC transport from the ER to Golgi. However, these are our speculations and more experiments are needed to further verify the above possibilities.
Materials and Methods
Generation of Dlic1-deficient mice
To construct the Dlic1 gene-targeting vector, we screened the 129/Sv mouse genomic phage library (Stratagene) and isolated a genomic DNA fragment containing Dlic1 exon 3 to exon 9. In the gene targeting vector, we inserted a FRT-flanked β-gal reporter and neo expression cassette at the NheI site before exon 5, and a Loxp-flanked NheI-SmaI fragment containing exon 5. A diphtheria toxin A (DTA) expression cassette was used as a negative selection marker in the targeting vector. The linearized Dlic1 targeting vector was electroporated into mouse embryo stem (ES) cells (W4/129S6 ES, Taconic Transgenic). After double selection, the recombinant ES cell clones were screened by PCR and further confirmed by Southern blot analysis with32P-labeled PvuII-BglII fragment containing exon 3. Two recombinant ES clones were injected into C57BL/6J blastocysts to produce chimera mice. The chimera mice were crossed with PGK-Cre transgenic mice to delete exon 5 and generate Dlic1+/galeo mice. Progenies were genotyped by PCR using primers: Dlic1SA: 5′-GGCAGAAACTCCTAACCAGCAAG-3′, Dlic1KO: 5′-GCTACTTATCCACAACCAACGCAC-3′ and Dlic1WT: 5′-CATACCTGAGACCCACAAAA CAGC-3′). Then Dlic1+/galeo mice were crossed with Act-Flp transgenic mice (003800, The Jackson Laboratory) to delete the β-gal and neo cassette to generate Dlic1+/− mice. Progenies were genotyped by PCR using primers: Dlic1LR: 5′-TGGCTCAGTGGTAAAGGTCC-3′, Dlic1LoxpR: 5′-TCAGGAAAAGCACTGG CTG-3′, Dlic1FRTF: 5′-GGAAGATGTGACAAGACAGACACG-3′. All of the Dlic1 mutant mice used for experiments were maintained on a C57BL/6 and 129/sv mixed genetic background. Mouse breeding and experimental manipulations were carried out following the general guidelines published by The Association for Assessment and Accreditation of Laboratory Animal Care. Animal-related procedures were reviewed and approved by the Animal Care and Use Committee of the Institute of Developmental Biology and Molecular Medicine at Fudan University.
Histological analysis and immunofluorescent staining
Frozen sections: after the animals were exterminated by cervical dislocation, eyes were dissected out and embedded in OCT. Samples were then quickly frozen in liquid nitrogen-cooled isopentane, and 10 μm cryosections were collected. For histological analysis, frozen sections were stained with hematoxylin and eosin for light microscopy. OS length and ONL thickness in retinas were measured in sections (on a Leica DMRXA2 microscope at 400× magnification). OS length was measured along the long axis of the OS between the inner segments and the retinal pigment epithelium. ONL thickness was analyzed by counting rows of photoreceptor cells in the outer nuclear layer. Five different areas in the central and midperipheral retina were measured and three mice for each genotype were used for statistics. Only representative images were used for statistical analysis. For immunofluorescent staining, the sections were stained with various antibodies as described previously45 except that the sections were fixed in 0.2% PFA for 10 min for immunofluorescent staining with anti-PCNT antibody. Fluorescence micrographs were acquired using a Leica DMRXA2 fluorescence microscope equipped with a Leica DFC350FX camera. Histological micrographs were acquired using a Leica DMRXA2 fluorescence microscope equipped with a Leica DFC300FX camera. Images were processed using Adobe Photoshop.
Statistical analysis
Statistical analyses were performed using Microsoft Excel 2007 and GraphPad Prism 4. The results are presented as mean ± SEM, and significant differences are indicated by a single asterisk * when P < 0.05 and a double asterisk ** when P < 0.01.
X-gal staining
Dlic1galeo/galeo mice were used for X-gal staining to determine the expression pattern of Dlic1. Eyes were fixed in 0.2% glutaraldehyde for 10 h followed by cryoprotection in 30% sucrose for overnight at 4 °C and then 20-μm frozen sections were collected. The X-gal staining was carried out following standard protocol46. The slices were subsequently counterstained with eosin.
TUNEL assays
TUNEL assays were performed as described previously47. For quantification, digital images were taken under fluorescence microscopy at 400× magnification, and the TUNEL-positive cells in the ONL of five randomly chosen fields in the central and midperipheral retina were counted for each retina. Three animals for each time point and each genotype were used for statistics.
Antibodies
A synthetic peptide consisting of the C-terminal 15 amino acids of the DLIC1 protein, VFPTTPTSPTEGEAS12, was used to raise polyclonal antibodies. Antibodies were affinity-purified with Hitrap NHS-activated HP columns (Amersham Biosciences). Antibodies against the following proteins were used for immunofluorescence: GFAP (MAB3402, Chemicon); arrestin (PA1-731, Affinity Bioreagent); fluorescein peanut agglutinin (L-1020, Vector); calnexin (SPA-860, Stressgen); DIC (74 kDa) (MAb1618, Millipore); DHC (SC-9115, Santa Cruz); giantin (A488-114L, Covance); GalT (generously provided by Dr Jack Rohrer); tctex-1 (11954-1-AP, Proteintech). Antibodies against Rhodopsin (R5403) and Ac-tubulin (T7451) were from Sigma; Rab11 (610656), p150 Glued (610474), and PCNT (611814) were from BD Bioscience. Secondary antibodies: goat anti-rabbit IgG-FITC (F9887) and goat anti-rabbit IgG-CY3 (C2306) were from Sigma; goat anti-mouse IgG-FITC (FI-2020) was from Vector Labs. All the secondary antibodies were diluted at 1:300 when used.
Immunoprecipitation and western blot analyses
Freshly dissected eyes were enucleated and two eye cups were homogenized in 200 μl TNP buffer48 to make retina protein extracts. Preparation of cell lysates from other tissues, co-immunoprecipitation and western blot analyses with antibodies against DIC, DHC, and DLIC1 were performed according to the procedures described previously48. For quantification of protein levels, appropriate film exposures were scanned, the density of bands were determined with image J and normalized to band intensity for GAPDH. Each experiment was repeated three times.
Quantitative PCR
Total RNA was extracted from mouse retinas using Trizol reagent (Invitrogen). cDNA was synthesized using RNA PCR kit (Takara) following the manufacturer's instructions. Quantitative PCR was carried out with 2× QuantiTect SYBR Green PCR master mix (Stratagen). The assay were repeated with five pairs of mice in two independent experiments. Values obtained for the target genes were calibrated with the housekeeping gene Gapdh. The mRNA levels of DHC and DIC were assessed as an increase or decrease in the mutant mice relative to control. Primers used were as follows: DIC-F1, 5′-AACCCCTATGTCTCCCTCTTCG-3′, DIC-R1, 5′- CGTCTCCCAAGTTCTGAGTCTGAC-3′, DHC-F1, 5′-GACAGGGATGGTGATAAGATGGC-3′, DHC-R1, 5′-TAACTTTGGGCTTTTC TCCGC-3′.
Electron microscopy
Eyes were enucleated and eye cups were fixed in the fixative buffer (0.05% glutaraldehyde plus paraformaldehyde in PBS, pH 7.4) at 4 °C for at least 24 h, and then embedded in resin. Ultrathin sections were prepared using a Reichert ultramicrotome, contrasted with uranyl acetate and lead citrate and examined under a Philips CM120 electron microscope.
Mouse electroretinograms
Mouse ERG was performed according to the procedure described previously with some minor modifications49. Mice were dark adapted overnight and anesthetized with a mixture of ketamine (15 mg/g body weight) and xylazine (5 mg/g body weight) under dim red light. The pupils were dilated with a single drop of 1% atropine sulfate. A drop of 0.5% proparacaine hydrochloride was applied for corneal anesthesia. A small amount of 2.5% methylcellulose gel was applied to the eye. Mice were placed on a heating pad keeping at 37 °C inside a Ganzfield dome (Roland Q400, Wiesbaden, Germany). A silver loop electrode was placed over the cornea to record the ERGs. Needle reference and ground electrodes were placed in the cheek and tail, respectively. The responses were differentially amplified (1-500 Hz). All stimuli were presented in the Ganzfeld dome. Light was spectrally filtered with a 500 nm interference filter. The intensities of flashes were -5.0, -15, -25 and -35 log scotopic candela-sec/m2 (cd. s/m2). For photopic ERG, mice were recovered at intensities of 30 cd. s/m2 for 10 min, and then tested at the intensity of 3.0 cd. s/m2. Data were collected and analyzed with Port32.exe . The mice tested were at the age of 10 months.
Cilium induction and length measurements
We prepared MEFs from the control and Dlic1−/− E13.5 embryos. Primary MEFs were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin-streptomycin (Invitrogen). To induce cilium growth, 3 × 105 cells were plated on a cover slide in a six-well plate, followed by serum starvation for 48 h to promote cilium growth. Then the cells were stained with anti-Ac-tubulin antibody to label the cilium.
To quantify the cilium lengths of Dlic1−/− photoreceptors and MEFs, fluorescent images of retinal sections or MEFs immunofluorescent-stained with anti-Ac-tubulin antibody from three independent experiments were acquired using a Leica DMRXA2 fluorescencse microscope at 400× (for MEFs) or 1 000× (for photoreceptors) magnification. Cilium in images were traced and measured with software (Image J version 1.46, NIH).
References
Young RW . The renewal of photoreceptor cell outer segments. J Cell Biol 1967; 33:61–72.
Tai AW, Chuang JZ, Bode C, Wolfrum U, Sung CH . Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell 1999; 97:877–887.
Wolfrum U, Schmitt A . Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil Cytoskeleton 2000; 46:95–107.
Ramamurthy V, Cayouette M . Development and disease of the photoreceptor cilium. Clin Genet 2009; 76:137–145.
Vallee RB, Williams JC, Varma D, Barnhart LE . Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol 2004; 58:189–200.
Pfister KK, Shah PR, Hummerich H, et al. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet 2006; 2:e1.
Tynan SH, Purohit A, Doxsey SJ, Vallee RB . Light intermediate chain 1 defines a functional subfraction of cytoplasmic dynein which binds to pericentrin. J Biol Chem 2000; 275:32763–32768.
Yoder JH, Han M . Cytoplasmic dynein light intermediate chain is required for discrete aspects of mitosis in Caenorhabditis elegans. Mol Biol Cell 2001; 12:2921–2933.
Lee WL, Kaiser MA, Cooper JA . The offloading model for dynein function: differential function of motor subunits. J Cell Biol 2005; 168:201–207.
Mische S, He Y, Ma L, Li M, Serr M, Hays TS . Dynein light intermediate chain: an essential subunit that contributes to spindle checkpoint inactivation. Mol Biol Cell 2008; 19:4918–4929.
Sivaram MV, Wadzinski TL, Redick SD, Manna T, Doxsey SJ . Dynein light intermediate chain 1 is required for progress through the spindle assembly checkpoint. EMBO J 2009; 28:902–914.
Bielli A, Thornqvist PO, Hendrick AG, Finn R, Fitzgerald K, McCaffrey MW . The small GTPase Rab4A interacts with the central region of cytoplasmic dynein light intermediate chain-1. Biochem Biophys Res Commun 2001; 281:1141–1153.
Horgan CP, Hanscom SR, Jolly RS, Futter CE, McCaffrey MW . Rab11-FIP3 links the Rab11 GTPase and cytoplasmic dynein to mediate transport to the endosomal-recycling compartment. J Cell Sci 2010; 123:181–191.
Palmer KJ, Hughes H, Stephens DJ . Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol Biol Cell 2009; 20:2885–2899.
Zhang J, Li S, Musa S, Zhou H, Xiang X . Dynein light intermediate chain in Aspergillus nidulans is essential for the interaction between heavy and intermediate chains. J Biol Chem 2009; 284:34760–34768.
Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML . Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci 2004; 24:3907–3916.
Satoh D, Sato D, Tsuyama T, et al. Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat Cell Biol 2008; 10:1164–1171.
Zheng Y, Wildonger J, Ye B, et al. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol 2008; 10:1172–1180.
Banks GT, Haas MA, Line S, et al. Behavioral and other phenotypes in a cytoplasmic Dynein light intermediate chain 1 mutant mouse. J Neurosci 2011; 31:5483–5494.
Dyer MA, Cepko CL . Control of Muller glial cell proliferation and activation following retinal injury. Nat Neurosci 2000; 3:873–880.
McLaren MJ . Kinetics of rod outer segment phagocytosis by cultured retinal pigment epithelial cells. Relationship to cell morphology. Invest Ophthalmol Vis Sci 1996; 37:1213–1224.
Knabe W, Kuhn HJ . Ciliogenesis in photoreceptor cells of the tree shrew retina. Anat Embryol (Berl) 1997; 196:123–131.
Dawe HR, Farr H, Gull K . Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J Cell Sci 2007; 120:7–15.
Avasthi P, Marshall WF . Stages of ciliogenesis and regulation of ciliary length. Differentiation 2012; 83:S30–42.
Muhlhans J, Brandstatter JH, Giessl A . The centrosomal protein pericentrin identified at the basal body complex of the connecting cilium in mouse photoreceptors. PLoS One 2011; 6:e26496.
Westlake CJ, Baye LM, Nachury MV, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA 2011; 108:2759–2764.
Knodler A, Feng S, Zhang J, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA 2010; 107:6346–6351.
Horgan CP, Hanscom SR, Jolly RS, Futter CE, McCaffrey MW . Rab11-FIP3 binds dynein light intermediate chain 2 and its overexpression fragments the Golgi complex. Biochem Biophys Res Commun 2010; 394:387–392.
Bachmann-Gagescu R, Phelps IG, Stearns G, et al. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet 2011; 20:4041–4055.
Satoh AK, O'Tousa JE, Ozaki K, Ready DF . Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Development 2005; 132:1487–1497.
Storrie B, White J, Rottger S, Stelzer EH, Suganuma T, Nilsson T . Recycling of golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J Cell Biol 1998; 143:1505–1521.
Nilsson T, Lucocq JM, Mackay D, Warren G . The membrane spanning domain of beta-1,4-galactosyltransferase specifies trans Golgi localization. EMBO J 1991; 10:3567–3575.
Kopito RR, Sitia R . Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep 2000; 1:225–231.
Insinna C, Baye LM, Amsterdam A, Besharse JC, Link BA . Analysis of a zebrafish dync1h1 mutant reveals multiple functions for cytoplasmic dynein 1 during retinal photoreceptor development. Neural Dev 2010; 5:12.
Yeh TY, Peretti D, Chuang JZ, Rodriguez-Boulan E, Sung CH . Regulatory dissociation of Tctex-1 light chain from dynein complex is essential for the apical delivery of rhodopsin. Traffic 2006; 7:1495–1502.
Rosenbaum JL, Witman GB . Intraflagellar transport. Nat Rev Mol Cell Biol 2002; 3:813–825.
Palmer KJ, MacCarthy-Morrogh L, Smyllie N, Stephens DJ . A role for Tctex-1 (DYNLT1) in controlling primary cilium length. Eur J Cell Biol 2011; 90:865–871.
Li A, Saito M, Chuang JZ, et al. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol 2011; 13:402–411.
Welte MA . Bidirectional transport: matchmaking for motors. Curr Biol 2010; 20:R410–413.
Welte MA . Bidirectional transport along microtubules. Curr Biol 2004; 14:R525–537.
Wozniak MJ, Bola B, Brownhill K, Yang YC, Levakova V, Allan VJ . Role of kinesin-1 and cytoplasmic dynein in endoplasmic reticulum movement in VERO cells. J Cell Sci 2009; 122:1979–1989.
Hughes H, Stephens DJ . Assembly, organization, and function of the COPII coat. Histochem Cell Biol 2008; 129:129–151.
Appenzeller-Herzog C, Hauri HP . The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 2006; 119:2173–2183.
Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ . Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol 2005; 7:48–55.
Yu J, Lei K, Zhou M, et al. KASH protein Syne-2/Nesprin-2 and SUN proteins SUN1/2 mediate nuclear migration during mammalian retinal development. Hum Mol Genet 2011; 20:1061–1073.
Nagy A . Manipulating the mouse embryo: a laboratory manual. 3rd Edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press 2003.
Tornusciolo DR, Schmidt RE, Roth KA . Simultaneous detection of TDT-mediated dUTP-biotin nick end-labeling (TUNEL)-positive cells and multiple immunohistochemical markers in single tissue sections. Biotechniques 1995; 19:800–805.
Zhang X, Lei K, Yuan X, et al. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 2009; 64:173–187.
Pennesi ME, Cho JH, Yang Z, et al. BETA2/NeuroD1 null mice: a new model for transcription factor-dependent photoreceptor degeneration. J Neurosci 2003; 23:453–461.
Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB945304, 2011CB510102, 2009CB526502, 2006CB806700), the National Natural Science Foundation of China (31171406, 30971666, 30630043), the National Hi-tech Research and Development Program of China (2007AA022101), Key Projects of Basic Research (08JC1400800) from the Science and Technology Committee of Shanghai Municipality, and the 211 and 985 projects of Ministry of Education of China. This work was also supported by Excellent PhD Student Research Grant of Fudan University to S Kong. We thank Dr Jack Rohrer for GalT antibody; Professor Tiande Shou for scientific discussion and technical help, Drs Yuan Zhuang, Min Han and Xiaohui Wu for scientific discussion and Mr Alan Yu for English editing.
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
(A and B) Statistical analysis of reduced body weight in both Dlic1−/− male (A) and female (B) mice. (PDF 2085 kb)
Supplementary information, Figure S2
Electron micrographs of outer segments of Dlic1+/− and Dlic1−/− photoreceptor cells show well packed disk membranes in both control and mutant OS. (PDF 4103 kb)
Supplementary information, Figure S3
Accumulation of rhodopsin in the ER of Dlic1−/− photoreceptor cells. (PDF 2249 kb)
Supplementary information, Figure S4
(A) Quantitative analysis of the reduction of DIC level in Dlic1−/− brain, liver and MEFs shown in Figure 6B. (PDF 275 kb)
Rights and permissions
About this article
Cite this article
Kong, S., Du, X., Peng, C. et al. Dlic1 deficiency impairs ciliogenesis of photoreceptors by destabilizing dynein. Cell Res 23, 835–850 (2013). https://doi.org/10.1038/cr.2013.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.59
Keywords
This article is cited by
-
Analysis of genome-wide knockout mouse database identifies candidate ciliopathy genes
Scientific Reports (2022)
