
Abstract
Best known for its role in targeting protein degradation by the proteasome, ubiquitin modification has also emerged as an important mechanism that regulates cell signaling through proteasome-independent mechanisms. The role of ubiquitin as a versatile signaling tag is characteristically illustrated in the NF-κB pathways, which regulate a variety of physiological and pathological processes in response to diverse stimuli. Here, we review the role of ubiquitination in different steps of the NF-κB signaling cascades, focusing on recent advances in understanding the mechanisms of protein kinase activation by polyubiquitin chains in different pathways that converge on NF-κB.
Similar content being viewed by others

The ubiquitin conjugation system
Believed to have evolved from ancestral proteins containing β-grasp folds 1, ubiquitin is a highly conserved and ubiquitously expressed protein in all eukaryotes from yeast to human. Four individual genes encode ubiquitin precursors either as a fusion protein between a ubiquitin and a ribosomal subunit or a “head-to-tail” linear polyubiquitin 2. The ubiquitin precursors are further processed into the 76-amino-acid mature protein, which contains a C-terminal tail that can be covalently attached to other proteins through a stepwise enzymatic reaction involving three classes of enzymes, E1, E2 and E3 (Figure 1) 3. The human genome encodes two E1s, ∼50 E2s and over 700 E3s, underscoring the enormous complexity of the ubiquitin system. In addition, similar to phosphorylation and dephosphorylation, ubiquitination can be reversed by deubiquitination enzymes (DUBs) that also comprise a large family of proteins. Moreover, bioinformatic and biochemical analyses have identified more than 20 types of ubiquitin-binding domains (UBDs), which are embedded in a large variety of proteins of diverse cellular functions 4. The recognition of ubiquitinated substrates by proteins containing UBDs forms a ubiquitin network that generates distinct functional outputs in response to different signals. In the most typical example, the recognition of polyubiquitinated proteins by the proteasome leads to the degradation of these proteins within the proteasome.

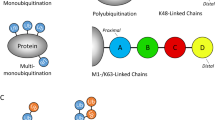
Regulation of protein functions by ubiquitination. (A) Schematic representation of the three-step ubiquitination cascade. Monoubiquitination and K63 polyubiquitination generally serve non-proteolytic functions, whereas polyubiquitin chains of other linkages target proteins for degradation by the proteasome. (B) Structure of ubiquitin (PDB code: 1UBQ), highlighting its seven lysine residues.
Ubiquitin has seven lysines (K6, K11, K27, K29, K33, K48 and K63), all of which can be conjugated to another ubiquitin to form a polyubiquitin chain (Figure 1). In addition, the amino terminus of one ubiquitin can be conjugated to the carboxyl terminus of another ubiquitin to form a linear polyubiquitin chain 5. Polyubiquitin chains synthesized through different lysine linkages serve distinct functions in the cell 6. For example, K48- and K11-linked polyubiquitin chains usually target proteins for proteasomal degradation, whereas K63 polyubiquitin chains and monoubiquitination regulate cellular functions such as protein kinase activation, DNA repair, membrane trafficking and chromatin remodeling, largely through proteasome-independent mechanisms 7, 8.
The NF-κB – IκB pathway
NF-κB is a family of heterodimeric transcription factors that regulate a large array of genes involved in immunity, inflammation and cell survival 9. The NF-κB family has five members, including p50, p52, p65 (RelA), c-Rel and RelB, all of which contain an N-terminal Rel homology domain (RHD), which mediates their dimerization, nuclear localization and DNA binding. The RHD also binds to inhibitory proteins of κB family (IκBs), which sequester NF-κB in the cytoplasm in unstimulated cells. A transcription activation domain (TAD) is present in p65, c-Rel and RelB to activate gene expression. The TAD is absent in p50 and p52, which are generated from their precursors, p105 and p100, respectively, by proteasomal degradation of the C-terminal IκB-like domain. p50 and p52 bind to a TAD-containing family member to generate a functional NF-κB dimer.
The IκB family consists of several ankyrin repeat-containing proteins, including IκBα, IκBβ, IκBɛ, IκB-δ (IκBNS), IκB-ζ and Bcl-3. IκBα, IκBβ and IκBɛ sequester NF-κB in the cytoplasm, whereas IκB-δ, IκB-ζ and Bcl-3 function in the nucleus as co-activators of NF-κB. The C-termini of p100 and p105 also contain IκB-like ankyrin repeats, which mimic the inhibitory function of IκB family members.
Ubiquitin-mediated protein degradation activates the canonical and noncanonical NF-κB pathways
The activation of NF-κB requires the degradation of IκB proteins or the processing of NF-κB precursors to the mature subunits 10. In most cases, IκB degradation is the hallmark of NF-κB activation, thus the signaling pathways that lead to IκB degradation are classified as the canonical NF-κB pathways (Figure 2, left). These pathways include those activated by cytokines such as tumor necrosis factor (TNF) α and interleukin (IL)-1β, and bacterial products such as lipopolysaccharides (LPS). Stimulation with any of these ligands leads to the activation of the IκB kinase (IKK) complex, which is composed of the catalytic subunits IKKα and IKKβ, and the essential regulatory subunit NEMO (also known as IKKγ or IKKAP). IKK phosphorylates IκB proteins and targets them for polyubiquitination by a ubiquitin ligase complex consisting of Skp1, Cul1, Roc1 and βTrCP 11. Ubiquitinated IκB is degraded by the 26S proteasome, allowing NF-κB to enter the nucleus to turn on target genes.

Role of ubiquitination in the canonical and noncanonical NF-κB pathways. In canonical NF-κB activation represented by IL-1R/TLR pathways (left), the receptors recruit the adaptor protein MyD88, the kinases IRAK4 and IRAK1 and the E3 ubiquitin ligase TRAF6. The recruitment causes TRAF6 oligomerization and activates its E3 ligase activity. Together with the E2 enzyme complex Ubc13/Uev1A, TRAF6 synthesizes unanchored K63-polyubiquitin chains that bind to the TAB2 and TAB3 subunits of the TAK1 kinase complex and the NEMO subunit of the IKK complex. This binding brings the kinases into proximity for phosphorylation and activation. IKK subsequently phosphorylates IκBα. Phosphorylated IκBα is recognized by the βTrCP E3 complex and targeted for ubiquitin-mediated proteasomal degradation, releasing the NF-κB dimer p50/p65 into the nucleus to turn on target genes including the IKK inhibitor A20. A20 forms a ubiquitin-editing complex with ITCH, RNF11 and TAX1BP1. One of the functions of this complex is to prevent K63 polyubiquitin chain synthesis by TRAF6 and Ubc13, which leads to IKK inhibition. In the noncanonical pathways (right), stimulation of the BAFF-R and CD40 on the B-cell surface leads to the recruitment of several E3 ligases, including TRAF2, TRAF3 and cIAPs. TRAF2 catalyzes K63 polyubiquitination of cIAPs, which in turn target TRAF3 for degradation by promoting its K48 polyubiquitination. In the absence of TRAF3, NIK is stabilized, leading to the activation of IKKα, which phosphorylates the NF-κB precursor p100. Phosphorylated p100 is also recognized by the βTrCP E3 complex and targeted for ubiquitin-mediated proteasomal processing to form the mature subunit p52. p52 forms a complex with Rel-B, which enters the nucleus to turn on genes that are important for B-cell activation and maturation.
In B lymphocytes, NF-κB can also be activated through a noncanonical pathway involving the processing of p100 to the mature p52 subunit by the proteasome 10 (Figure 2, right). In response to stimulation of a subset of receptors on B cells, such as CD40 and B-cell activating factor receptor (BAFF-R), IKKα is activated to phosphorylate p100. This phosphorylation leads to polyubiquitination of p100 by the βTrCP ubiquitin ligase complex. Interestingly, ubiquitination of p100 does not result in its complete degradation by the proteasome. Instead, the 26S proteasome selectively degrades the C-terminal ankyrin repeats of p100, leaving the N-terminus (p52) intact. This partial degradation of p100 by the proteasome is likely due to a glycine-rich region in p100 that prevents the proteasome from degrading the N-terminus 12. In addition, the tightly folded N-terminal RHD domain, which forms a stable dimer, may be resistant to degradation by the proteasome 13.
Ubiquitin-mediated protein kinase activation in diverse NF-κB signaling pathways
Since phosphorylation of IκBs and p100 by IKK is a prerequisite for their degradation or processing, it is crucial to understand how IKK is activated by different signal transduction pathways. Interestingly, ubiquitin is key to IKK regulation, but in a manner that does not involve proteasomal degradation 14. The first clue that ubiquitination may regulate IKK was provided by biochemical fractionation experiments that identified the ∼700 kDa IKK complex, which was later shown to contain three distinct IKK subunits 15. Unexpectedly, it was found that the IKK complex was activated by a polyubiquitination reaction catalyzed by ubiquitin E1 and an E2 of the Ubc4/5 family, and that this activation was independent of the proteasome and K48 of ubiquitin. These results suggest a proteasome-independent mechanism of IKK activation by polyubiquitination, but the physiological relevance of this finding remained obscure until TRAF6, a protein known to be essential for NF-κB activation by the IL-1 and Toll-like receptor (IL-1R/TLR) pathways 16, 17, was shown to function as a ubiquitin ligase 18. Through its N-terminal RING domain, which is highly conserved among all but one TRAF proteins, TRAF6 facilitates the synthesis of K63 polyubiquitin chains by the ubiquitin E2 complex Ubc13/Uev1A. This polyubiquitination leads to the activation of IKK. In recent years, much progress has been made in understanding the mechanism of IKK regulation by ubiquitination and deubiquitination. Furthermore, studies of different NF-κB signaling pathways reveal that ubiquitin-mediated activation of protein kinases is a common mechanism underlying NF-κB regulation by diverse pathways. The following sections will focus on the regulation of protein kinases by ubiquitination in different pathways, and the accompanying review by Harhaj and Dixit will discuss regulation of NF-κB by deubiquitination.
IL-1R/TLR signaling pathways
Interluekin-1 (IL-1) is a group of cytokines that are secreted by cells during inflammation as a defense and healing response to harmful stimuli 19. Upon binding their receptors on the plasma membrane, both IL-1α and IL-1β potently activate downstream signaling pathways that lead to the expression of a plethora of pro-inflammatory genes (Figure 2, left). TLRs are pathogen recognition receptors that detect specific molecular patterns of microbes such as bacteria and viruses. IL-1R and TLR share a similar cytosolic domain termed the Toll-IL-1R (TIR) domain, which recruits other TIR domain-containing adaptors such as myeloid differentiation primary gene 88 (MyD88). MyD88 in turn recruits two IL-1 Receptor-associated kinases, IRAK4 and IRAK1. IRAK1 binds to TRAF6, which then functions together with Ubc13/Uev1A to catalyze K63 polyubiquitination, leading to the activation of a protein complex consisting of the kinase TGF-β activated kinase 1 (TAK1; also known as mitogen-activated protein kinase kinase kinase 7) and the adaptor proteins TAB1 and TAB2 20, 21. Both TAB2 and its homolog TAB3 contain an NZF-type UBD that binds preferentially to K63 polyubiquitin chains, and this binding leads to autophosphorylation and activation of TAK1 22. Subsequently, the TAK1 complex phosphorylates IKKβ, leading to the activation of IKK and NF-κB. TAK1 also phosphorylates other kinases including MKKs, which activate the JNK and p38 kinase cascades.
Although the model of ubiquitin-mediated activation of protein kinases is supported by multiple lines of evidence from different labs, two early studies initially casted some doubts over this model. In one study, deletion of the RING domain of TRAF6 was found to block the activation of TAK1 and JNK but not of IKK by IL-1β 23. However, subsequent studies by several labs using TRAF6-deficient cells led to the consistent conclusion that the RING domain of TRAF6 is essential for IKK activation in the IL-1 pathway 24, 25, 26, 27, 28, 29. In another study, knockout of Ubc13 was found to impair JNK and p38 kinase activation, but IKK activation was largely normal 30. Follow-up studies, however, showed that Ubc13 and its catalytic activity are indeed essential for IKK activation by IL-1β and other cytokines 29, 31, 32, 33. The discrepancy is likely caused by incomplete depletion of Ubc13 by Cre recombinase in the earlier study. Indeed, by using different titers of Cre retroviruses which resulted in varying degrees of Ubc13 depletion, Yamazaki et al. 29 demonstrated that complete depletion of Ubc13 blocked IKK activation, whereas even a small residual amount of Ubc13 was sufficient to support normal IKK activation. More direct evidence that K63 polyubiquitination is required for IKK activation in cells was obtained through a strategy that allows tetracycline-inducible replacement of endogenous ubiquitin with the K63R mutant 32. In cells that are devoid of K63 polyubiquitination, IKK activation by IL-1β is completely blocked. Interestingly, these studies also reveal that IKK activation by TNFα involves a ubiquitin-dependent mechanism that is not restricted to K63 of ubiquitin (see below).
The mechanism by which TAK1 and IKK are activated by TRAF6-mediated polyubiquitination has been a subject of intense investigation. Early studies showed that several proteins in the IL-1 pathway are ubiquitinated following stimulation of cells. These include TRAF6, IRAK1, IRAK2, TAK1, TAB2 and NEMO 21, 24, 34, 35, 36. Evidence has been presented to support the role of ubiquitination of each of these proteins in IKK activation. However, conflicting results have also been reported. For example, complementation of TRAF6-deficient MEF cells with a lysine-less TRAF6 mutant restores normal IKK activation by IL-1β, suggesting that ubiquitination of TRAF6 may be dispensable in this pathway 28. Recent biochemical studies reveal that free K63 polyubiquitin chains, which are not conjugated to any cellular protein, can directly activate the TAK1 kinase complex 37. These polyubiquitin chains, which are synthesized by TRAF6 and Ubc13/Uev1A, associate with the UBD of TAB2 (and possibly TAB3). This binding promotes the autophosphorylation of TAK1 at Thr-187, resulting in its activation. TAK1 then phosphorylates and activates IKKβ in a manner that depends on NEMO.
While the crucial role of NEMO in IKK activation is well established, how NEMO regulates IKK activation is not well understood. Recent studies have suggested that the regulatory function of NEMO is intimately linked to its ability to bind polyubiquitin chains and/or its modification by ubiquitin 38. NEMO contains two UBDs: a coiled-coil domain (known as NUB, NOA, UBAN or CoZi) and a C-terminal zinc finger (ZnF) domain 39, 40, 41, 42. The NUB domain binds both linear and K63 polyubiquitin chains 40. Biochemical and structural studies reveal that the NUB domain binds to short linear ubiquitin chains with greater affinity than to short K63 chains 43, 44. However, a NEMO fragment containing both NUB and the C-terminal ZnF domains binds to both types of ubiquitin chains with similar affinity 45. Mutations that disrupt the binding of NEMO to ubiquitin chains also impair IKK activation 39, 40, 43. Importantly, several ubiquitin-binding mutations of NEMO have been found in patients suffering from ectodermal dysplasia with immunodeficiency, underscoring the important role of NEMO ubiquitin binding in NF-κB-mediated immune response 46.
Several studies suggest that ubiquitination of NEMO is also important for IKK activation. Tang et al. 47 provided the first evidence suggesting that NEMO is polyubiquitinated by cellular inhibitor of apoptosis protein (cIAP) 1 in response to TNFα stimulation. Subsequently, studies of different NF-κB signaling pathways revealed several potential ubiquitination sites on NEMO (e.g., K285, K277, K309 and K399) 48, 49, 50, 51, 52, 53. Different types of ubiquitin modifications on NEMO have been reported; these include monoubiquitination as well as polyubiquitination by K6, K63 or linear ubiquitin chains. Further study is needed to clarify the role of NEMO ubiquitination in IKK activation in different pathways.
The role of ubiquitination in regulating NF-κB is further underscored by the findings that several DUBs inhibit TAK1 and IKK by cleaving K63 polyubiquitin chains or preventing polyubiquitin chain synthesis. For example, the familial cylindromatosis tumor suppressor (CYLD) is a DUB that can specifically cleave K63 polyubiquitin chains 34, 54, 55. A20, another DUB that has potent anti-inflammatory activity, not only disassembles polyubiquitin chains but also prevents polyubiquitin chain synthesis by blocking the interaction between TRAF6 and Ubc13 56, 57, 58, 59.
TNF receptor (TNFR) pathways
TNF is a potent inflammatory cytokine and it can also induce apoptosis of certain tumor cells 60. There are two forms of TNFα, a membrane-bound form (mTNFα) and a soluble form (sTNFα), the latter being produced by the processing of mTNFα by the membrane-bound protease TNFα converting enzyme (TACE). Both forms are functionally active to bind TNF receptors 61. Two TNF receptors, TNFR1 and TNFR2, bind TNFα, with TNFR1 playing a more dominant role in TNFα signaling. Upon binding to TNFα, which is a trimer, TNFR1 also forms a trimer that recruits TNFR1-associated death domain protein (TRADD) and triggers the sequential formation of two distinct complexes: complex I and II, which activate NF-κB and apoptosis, respectively (Figure 3) 62.

Role of ubiquitination in TNFα-induced NF-κB activation and apoptosis. Binding of TNFα to its receptor (TNFR1) leads to the assembly of a membrane receptor complex (complex I) by recruiting TRADD, TRAF2 or TRAF5, RIP1 and cIAPs. HOIP and HOIL-1 may also help to stabilize complex I by binding to polyubiquitin chains on cIAPs. RIP1 is polyubiquitinated by cIAPs and this modification leads to the recruitment of the TAK1 and IKK complexes. Subsequently, IκBα is phosphorylated and degraded, leading to NF-κB activation. Following complex I formation, TRADD, TRAF2 and RIP1 dissociate from TNF receptor and form a death-inducing complex with FADD and procaspase-8 (complex II) in the cytosol. This allows procaspase-8 to undergo autocatalytic cleavage to generate the mature caspase-8, which initiates apoptosis. However, apoptosis is normally prevented by NF-κB-induced expression of anti-apoptotic proteins such as c-FLIP, which prevents caspase-8 activation. Apoptosis is promoted by ITCH and SMAC, which target c-FLIP and cIAPs, respectively, for proteasomal degradation. NF-κB also induces A20, which forms a ubiquitin-editing complex that targets RIP1 for proteasomal degradation.
Complex I contains TRADD, TRAF2, TRAF5, receptor-interacting protein kinase 1 (RIP1), and cIAP1 and cIAP2. Both cIAP1 and cIAP2 are RING-domain ubiquitin ligases that catalyze polyubiquitination of RIP1 32, 63, 64. The polyubiquitin chains facilitate the recruitment and activation of TAK1 and IKK complexes by binding to TAB2 and NEMO, respectively 39. Subsequent to the formation of the receptor-associated complex I and the activation of TAK1, IKK and NF-κB, TRADD, RIP1 and TRAF2 dissociate from the receptor to form complex II in the cytoplasm together with FADD and procaspase-8 62. Within complex II, procaspase-8 is processed into the mature caspase-8, which then cleaves and activates caspase-3 to trigger apoptosis. In most cells, however, TNFα does not induce apoptosis, because the rapid activation of NF-κB by complex I induces the synthesis of several anti-apoptotic proteins including cIAPs and c-FLIP. c-FLIP binds to procaspase-8 and prevents its activation. Inhibition of RIP1 polyubiquitination, by mutating its ubiquitination site (K377), promoting cIAPs degradation or deubiquitinating RIP1, leads to caspase-8 activation and apoptosis 58, 65, 66.
The TNFα and IL-1 signaling pathways share some similarities and apparent differences. Both pathways activate TAK1 and IKK through TRAF proteins (TRAF2 and 5 for TNFα and TRAF6 for IL-1β). In addition, both pathways can be inhibited by DUBs such as CYLD and A20. These similarities led to the early suggestions that both pathways activate TAK1 and IKK through a mechanism involving K63 polyubiquitination. Indeed, TNFα-induced K63 polyubiquitination of RIP1 has been directly observed using a K63-specific antibody 67. However, several lines of evidence suggest that TNFα and IL-1β activate IKK through distinct ubiquitin-dependent mechanisms 32. First, depletion of Ubc13 blocks IKK activation by IL-1β, but not TNFα. Second, replacement of endogenous ubiquitin with the K63R ubiquitin mutant prevents IKK activation by IL-1β, but not TNFα. Third, depletion of Ubc5 inhibits IKK activation by TNFα but not IL-1β. Fourth, cIAP1 and cIAP2 are required for RIP1 polyubiquitination and IKK activation in response to TNFα stimulation. Thus, unlike Ubc13 and TRAF6 that catalyze K63 polyubiquitination in the IL-1β pathway, the TNFα pathway employs cIAPs and Ubc5 to catalyze polyubiquitination of RIP1 that is not restricted to K63 of ubiquitin. Interestingly, the TNF receptor complex contains HOIP and HOIL-1, which form the LUBAC ubiquitin E3 complex capable of synthesizing linear polyubiquitin chains 5, 48, 68. The recruitment of LUBAC to TNFR requires TRADD, TRAF2 and cIAP1/2, but not RIP1 or NEMO. RNAi of HOIP partially inhibits IKK activation by TNFα. These results, together with the findings that LUBAC promotes linear polyubiquitination of NEMO and that NEMO binds to linear polyubiquitin chains, suggest that linear polyubiquitination may be important for IKK activation by TNFα. However, so far there is no evidence that linear polyubiquitin chains are associated with endogenous NEMO in cells. In addition, there is no direct biochemical evidence that linear ubiquitination of NEMO or its binding to linear ubiquitin chains leads to IKK activation. Thus, further studies are needed to elucidate the precise mechanism of IKK activation in the TNFα pathway.
T-cell receptor pathways
T-cell receptors (TCRs) are activated by antigenic peptides presented by host major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs) 69, 70. TCRs subsequently lead to the activation of the serine/threonine kinase PKCθ by initiating a cascade of intracellular protein tyrosine phosphorylation. PKCθ then recruits a protein complex consisting of the CARD domain proteins CARMA1, BCL10 and a caspase-like protein MALT1. This complex, named as CBM (CARMA1-BCL10-MALT1), activates IKK through a mechanism involving K63 polyubiquitination by Ubc13/Uev1A (Figure 4, upper left). Indeed, deletion of Ubc13 in mouse T cells prevents the activation of TAK1, IKK and MAP kinases by TCR stimulation 31. In addition, MALT1 contains binding sites for TRAF2 and TRAF6. Biochemical studies show that BCL10 and MALT1 promote the oligomerization of TRAF6 and activate its E3 ligase activity 35. TRAF6, together with Ubc13/Uev1A, in turn activates TAK1 and IKK. It has also been suggested that MALT1 harbors an E3 ligase activity, which functions together with Ubc13/Uev1A to catalyze K63 polyubiquitination of NEMO 50. Although K399 of NEMO has been shown to be polyubiquitinated in response to TCR stimulation, mouse “knock-in” experiments show that this lysine is dispensable for NF-κB activation in T cells 71. Other targets shown to be polyubiquitinated by TRAF6 include BCL10 and MALT1 72, 73. However, T-cell-specific deletion of TRAF6 in mice does not impair NF-κB activation by TCR 74. This may be due to redundant functions of TRAF2 and TRAF6 in T cells, as RNAi of both genes, but not of either one alone, abrogates NF-κB activation by TCR 35.

Expanding role of ubiquitination in protein kinase activation in diverse NF-κB pathways. TCR pathway (upper left): Upon TCR stimulation, the serine/threonine kinase PKCθ is activated by a tyrosine kinase cascade. PKCθ promotes the formation of a protein complex composed of CARMA1, BCL10 and MALT1. MALT1 recruits TRAF2 and TRAF6, which catalyze K63 polyubiquitination that leads to the activation of TAK1 and IKK. NLR pathway (upper center): In response to bacterial peptidoglycans, cytosolic receptors NODs recruit TRAF proteins and target RIP2 for K63 ubiquitination, leading to the activation of TAK1 and IKK. RLR pathway (upper right): After viral infection, the viral RNA binds to the cytosolic RNA helicase RIG-I, which then binds to unanchored K63 polyubiquitin chains synthesized by the ubiquitin ligase TRIM25. This binding activates RIG-I, which transduces the signal to MAVS, a mitochondrial membrane protein. MAVS in turn activates IKK and TBK1, leading to the activation of NF-κB and IRF3, respectively. NF-κB and IRF3 enter the nucleus to induce the transcription of type-I interferons (IFN-Is). DNA damage pathway (bottom): Genotoxic stress caused by DNA double-strand breaks triggers the sequential modification of NEMO by SUMO1 and ubiquitin. Monoubiquitinated NEMO exits the nucleus together with ATM and this complex promotes K63 polyubiquitination of ELKS and/or TRAF6, which in turn activates TAK1 and IKK.
The TCR pathway is subjected to negative regulation in part through ubiquitination and deubiquitination. The ubiquitin ligase Cbl-b, a well known negative regulator of TCR signaling, can promote monoubiquitination of CARMA1 to disrupt the CBM complex, thereby inhibiting NF-κB 75, 76. T cells lacking the DUB CYLD exhibit constitutive activation of TAK1 and IKK, and this is correlated with enhanced ubiquitination of TAK1 77. It also has been suggested that A20 inhibits NF-κB in T cells by removing K63 ubiquitin chains from MALT1 78, 79. Interestingly, MALT1 possesses protease activity that can cleave A20 and BCL10; the cleavage of A20 may contribute to the requirement of the MALT1 protease activity in optimal NF-κB activation and IL-2 production 80, 81. The contribution of the MALT1 protease activity to NF-κB activation in lymphocytes is further supported by the observation that inhibition of MALT1 proteolytic activity suppressed cancer cell growth that depends on constitutive NF-κB activity 82, 83.
RIG-I-like receptor (RLR) pathways
Upon infection by RNA viruses, viral RNA represents a pathogen-associated molecular pattern (PAMP) that alerts the host immune system 84, 85. The viral RNA in the cytosol is recognized by members of the RLR family, including retinoic acid-inducible gene-I (RIG-I), melanoma differentiation-associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2) 86. All RLRs contain an RNA helicase domain, and RIG-I and MDA5 additionally contain two CARD domains at their N-termini, which are important for activating downstream signaling cascade that culminates in the induction of type-I interferons and other antiviral molecules (see below). RIG-I also contains a C-terminal regulatory domain (RD) that binds to RNA bearing 5′-triphosphate 87, 88. Genetic studies have demonstrated that RIG-I is essential for innate immune defense against most RNA virus infections, whereas MDA5 specifically detects picornaviruses such as encephalomyocarditis virus (EMCV) 85, 89. The specific ligand(s) that activates MDA5 is still not defined, but long double-stranded RNA such as poly[I:C] is known to induce interferons in an MDA5-dependent manner. LGP2 lacks the N-terminal CARD domains and was thought to inhibit the antiviral response 90. However, LGP2-deficient mice exhibit an impaired immune response to several RNA viruses, especially EMCV, suggesting that LGP-2 is a positive regulator of MDA5 and possibly RIG-I 91.
Binding of viral RNA to RIG-I and MDA5 initiates a signaling cascade through the adaptor protein MAVS (also known as IPS-1, VISA or Cardif) 92, 93, 94, 95. MAVS is predominantly localized on the mitochondrial outer membrane through a C-terminal transmembrane domain, and this localization is essential for its ability to induce type-I interferons 92. MAVS contains an N-terminal CARD domain that interacts with the CARD domains of RIG-I. The interaction with RIG-I leads to the activation of MAVS, which in turn activates IKK in the cytosol, leading to NF-κB activation. MAVS also activates the cytosolic IKK-related kinases TBK1 and IKKɛ, which phosphorylate the transcription factor IRF3. Phosphorylated IRF3 forms a dimer and enters the nucleus, where it functions together with NF-κB and other transcription factors to form an enhanceosome complex to induce type-I interferons (Figure 4, upper right).
Recent studies provide compelling evidence for a critical role of K63 polyubiquitination in signal transduction through different steps of the RIG-I antiviral pathway. Tripartite motif-25 (TRIM25), an interferon-inducible E3 ligase, was first shown to activate RIG-I by conjugating K63-linked polyubiquitin chains to K172 of RIG-I 96. The mechanism of RIG-I activation was further delineated using a cell-free system that recapitulates the activation of the RIG-I pathway by viral RNA 97. Surprisingly, RIG-I is activated by the binding of its N-terminal CARD domains to unanchored K63 polyubiquitin chains. These ubiquitin chains exist in human cells but they are very unstable due to abundant DUBs such as CYLD. However, the endogenous ubiquitin chains are very potent activators of RIG-I. The CARD domains of RIG-I bind to K63 ubiquitin chains selectively, and this binding in full-length RIG-I requires ATP and RNA. Mutations that disrupt the binding of RIG-I to K63 ubiquitin chains also abrogate its ability to induce type-I interferons. On the other hand, a mutation that disrupts ubiquitination of RIG-I but retains its ubiquitin binding does not impair its ability to induce type-I interferons, suggesting that ubiquitin-binding rather than ubiquitination of RIG-I leads to its activation. Thus, the activation of RIG-I requires its sequential binding of two ligands, viral RNA followed by K63 polyubiquitin chains. How polyubiquitin binding leads to the activation of RIG-I and subsequent activation of MAVS requires further research.
Following its activation by RIG-I, MAVS activates IKK and TBK1 also through a ubiquitin-dependent mechanism. Biochemical studies show that the ubiquitin E2 Ubc5 is necessary for TBK1 activation by MAVS in an in vitro assay 98. Moreover, depletion of Ubc5 abrogates TBK1 activation by viral infection in cells. Interestingly, although Ubc5 can synthesize polyubiquitin chains of different linkages, only K63 polyubiquitin chains can support TBK1 activation by MAVS. Replacement of endogenous ubiquitin with the K63R ubiquitin mutant in a human cell line leads to a severe defect in TBK1 activation by viral infection. MAVS contains binding sites for several TRAF proteins, including TRAF2, TRAF3, TRAF5 and TRAF6. TRAF3 forms a complex with TANK, NEMO and TBK1 98, 99, 100. NEMO is required for the activation of both IKK and TBK1 in response to viral infection, and mutations that disrupt the binding of NEMO to polyubiquitin chains impair the activation of both kinases 98, 100. Although autoubiquitination of TRAF3 has been proposed as a mechanism that leads to TBK1 activation, TRAF3-deficient cells only exhibit a partial defect in producing IFNβ following viral infection 98. Thus, additional ubiquitin E3 and ubiquitination targets involved in TBK1 activation remain to be identified. Further research is also needed to understand how IKK is activated by MAVS.
In support of the role of polyubiquitination in the activation of the RIG-I pathway, several DUBs have been shown to inhibit IKK and TBK1 activation by viral infection. These include A20, CYLD and DUBA 101, 102, 103, 104. A20 inhibits the RIG-I pathway at a step upstream of IKK and TBK1. However, this inhibition is independent of A20's catalytic activity. Thus, the mechanism by which A20 inhibits IKK and TBK1 activation is still not well understood. CYLD has been shown to deubiquitinate or prevent ubiquitination of RIG-I in cells. DUBA is an OTU-type DUB and it inhibits TBK1, but not IKK activation by removing polyubiquitin chains from TRAF3.
NOD-like receptor (NLR) pathways
Nucleotide-binding domain (NBD), leucine-rich repeat (LRR)-containing proteins (NLR; also known as NOD-like receptors) are cytosolic proteins that detect PAMPs or damage-associated molecular patterns (DAMPs) 105, 106. As their name implies, NLR proteins contain NBD and LRR domains. In addition, NLR proteins are classified into different subfamilies depending on whether they possess CARD, pyrin or baculovirus inhibitor of apoptosis protein repeat (BIR) domains. Among about 20 different NLR proteins, NOD1 and NOD2, which activate NF-κB, are among the most extensively studied 107, 108, 109.
Both NOD1 and NOD2 recognize bacterial peptidoglycans. NOD1 senses the dipeptide γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), whereas NOD2 detects muramyl dipeptide (MDP). RIP2, a RIP1-like kinase (also known as RICK) is recruited to NODs through CARD-CARD interaction upon ligand stimulation 107 and is subsequently polyubiquitinated at K209 within its kinase domain 110, 111. Polyubiquitinated RIP2 in turn recruits and activates TAK1 and IKK complexes. It has also been shown that NEMO is ubiquitinated at K285 after NOD2 stimulation 51. Mutations of NOD2 have been linked to Crohn's disease, and several of these mutations impair the ability of NOD2 to stimulate NEMO ubiquitination (Figure 4, upper center).
Multiple E3s have been reported to catalyze the ubiquitination of NEMO and/or RIP2. TRAF6 has been shown to be involved in the K63 polyubiquitination of NEMO as well as RIP2 in response to NOD2 stimulation 51, 111. However, another study showed that depletion of TRAF2 and TRAF5, but not TRAF6, ablated NF-κB activation in response to a NOD1 ligand 110. More recently, two separate studies found that cIAPs (cIAP1 and 2) and X-linked inhibitor of apoptosis (XIAP; XIAP1 and 2) bind to RIP2 and are important for NF-κB activation in NOD signaling 112, 113. Furthermore, cIAP1 and 2 were suggested as the direct E3 ligases that catalyze K63 polyubiquitination of RIP2 113. Thus, the NOD-RIP2 and TNFR-RIP1 pathways appear to activate IKK through a similar mechanism that involves polyubiquitination of RIP proteins by cIAP and TRAF E3s. Also, similar to the TNF pathway, A20 negatively regulates NF-κB activation by NOD1 and NOD2, possibly by inhibiting RIP2 polyubiquitination 110, 114.
NF-κB activation by genotoxic stress
Genotoxic stress induces NF-κB activation in a unique way because the DNA damage signals originate in the nucleus and IKK activation occurs in the cytoplasm. A signaling pathway from the nuclear DNA damage to cytosolic IKK activation has been revealed (Figure 4, bottom) 115. Interestingly, this pathway involves sequential posttranslational modifications of NEMO, including sumoylation and ubiquitination 49. Upon DNA damage, a small fraction of NEMO (free from IKKα/β) is conjugated in the nucleus by SUMO-1 at lysines 277 and 309. The E2 and E3 responsible for NEMO sumoylation are Ubc9 and protein inhibitor of activated STATy (PIASy), respectively 116. NEMO sumoylation requires p53-induced protein with a death domain (PIDD), RIP1 and poly (ADP-ribose) polymerase 1 (PARP-1) 117. In particular, PARP-1 detects DNA damage and catalyzes poly-ADP-ribosylation (PAR) of PARP-1 itself. The ADP-ribose polymer then binds to a PAR-binding motif (PARBM) present in PIASy, which then sumoylates NEMO 118. PARP1 also recruits ATM, a nuclear protein kinase activated by DNA double-strand breaks (DSBs), to phosphorylate NEMO at Ser85 119. Phosphorylated NEMO is subsequently monoubiquitinated and it enters the cytosol together with ATM. The NEMO-ATM complex promotes the K63 polyubiquitination of the adaptor protein ELKS by XIAP and Ubc13, leading to the activation of the TAK1 kinase complex 120. TAK1 then phosphorylates IKK, resulting in NF-κB activation. An independent study confirms the important role of Ubc13 and TAK1 in IKK activation by DNA damage, but finds that K63 polyubiquitination of TRAF6 is important for TAK1 activation 53. This study also shows that NEMO is monoubiquitinated at K285 in the cytoplasm by cIAP1 and that this ubiquitination is important for IKK activation.
NF-κB activation by viral proteins
Human T-lymphotropic virus type I (HTLV-1) is a retrovirus that infects T cells and causes adult T-cell leukemia. The Tax protein of the virus is a potent activator of IKK and NF-κB 121. Tax is ubiquitinated in a Ubc13-dependent manner 122. Ubiquitinated Tax recruits and activates the TAK1 and IKK complexes 123, 124. Overexpression of TRAF2, TRAF5 or TRAF6 strongly induces Tax ubiquitination, but it is unclear which of these TRAF proteins is the physiological E3 for Tax. A Tax-binding protein, TaxBP1, is a component of the A20 ubiquitin-editing complex 125. Through binding to TaxBP1, Tax disrupts the A20 complex and inhibits its negative regulation of IKK 126. The inhibition of A20 by Tax may contribute to the constitutive activation of NF-κB in HTLV-1-infected cells.
Proteins from other viruses have been shown to activate NF-κB as well. For instance, the latent membrane protein 1 (LMP1) of Epstein-Barr virus (EBV) and Saimiri transforming protein c (STP-c) from herpesvirus saimiri (HVS) both activate IKK through TRAF6 127, 128, 129. Some viral proteins target negative regulators of IKK to augment NF-κB activation induced by stress conditions, thereby promoting the survival and transformation of cancer cells. For example, the E6 protein of human papillomavirus (HPV) targets CYLD for ubiquitination and proteasomal degradation, leading to persistent NF-κB activation by hypoxia in HPV-infected cancer cells 130.
Noncanonical NF-κB signaling
The protein kinase nuclear factor κB-inducing kinase (NIK) is important for the processing of p100 to p52 in the noncanonical NF-κB pathway (Figure 2, right) 131. In B cells, NIK is constitutively degraded through the ubiquitin-proteasome system 132. Stimulation of B-cell surface receptors such as BAFF-R or CD40 leads to the stabilization of NIK, which in turn phosphorylates and activates IKKα, leading to the phosphorylation and subsequent processing of p100 by the proteasome 133. TRAF3 plays an important role in the degradation of NIK in B cells; however, TRAF3 is not the direct ubiquitin ligase for NIK. Instead, TRAF3 functions as an adaptor that recruits cIAP1 and cIAP2, which polyubiquitinate NIK and target the kinase for degradation by the proteasome 134. Stimulation of CD40 activates the E3 ligase activity of TRAF2, which catalyzes K63 polyubiquitination of cIAPs 135, 136. Through an unknown mechanism, K63 polyubiquitinated cIAPs switch their target from NIK to TRAF3, causing K48 polyubiquitination and subsequent degradation of TRAF3. The degradation of TRAF3 allows NIK to accumulate in activated B cells.
Conclusions and perspectives
A wealth of evidence supports the key role of ubiquitination in regulating signal transduction in the NF-κB pathways. It is now clear that ubiquitination not only controls the degradation of IκBs and the processing of NF-κB precursors (p100) by the proteasome, but also regulates the activation of IKK through proteasome-independent mechanisms. It is remarkable to note that many proteins involved in the regulation of IKK have ubiquitin-related functions, including ubiquitin conjugation (e.g., TRAFs, cIAPs and Ubcs), deconjugation (e.g., A20 and CYLD) and recognition (e.g., NEMO and TAB2). Ubiquitination also regulates the activation of MAP kinases (e.g., TAK1, JNK and p38) and TBK1, suggesting that ubiquitin has a more general role in regulating protein kinase activation in immune and inflammatory pathways.
The basic mechanism underlying ubiquitin signaling involves the interaction between ubiquitinated targets and ubiquitin receptors that harbor UBDs. This mechanism applies to both signaling to the proteasome and proteasome-independent functions of ubiquitin. The 19S regulatory particle of the proteasome contains several ubiquitin receptor subunits that recognize polyubiquitinated proteins destined for degradation 137. As cells contain numerous proteins that are ubiquitinated and perhaps even more proteins that contain UBDs, how is a ubiquitinated protein sorted out and targeted to the proteasome or other destinations to perform specific regulatory functions? It is generally believed that different polyubiquitin chains on a protein target determine whether a protein is degraded by the proteasome or not. For example, K48 polyubiquitin chains target proteasomal degradation, whereas K63 polyubiquitination performs non-proteolytic functions. In support of this model, quantitative mass spectrometry shows that inhibition of the proteasome in yeast cells leads to the accumulation of ubiquitin chains of different linkages, except K63 linkage 138. However, in vitro experiments show that K63 polyubiquitinated proteins are efficiently degraded by the 26S proteasome 139. The inefficient degradation of K63 polyubiquitinated proteins in vivo may be due to their sequestration by other ubiquitin-binding proteins in cells, as well as their faster deconjugation by the proteasome-associated DUBs 140.
In general, UBDs bind to ubiquitin and ubiquitin chains with low affinity and selectivity. Thus, the specificity of sorting different ubiquitinated proteins for different fates cannot rely on the sole interaction between ubiquitin and UBDs. Additional interactions between other domains of the ubiquitinated proteins and ubiquitin receptors can greatly enhance the specificity. Space and time must also play a key role in determining the specificity of ubiquitin signaling. Since ubiquitin chains are very labile due to abundant DUBs in cells, the “life time” of a ubiquitin chain on a given substrate is likely to be quite short. Thus, the interaction between a ubiquitinated protein and a ubiquitin receptor must occur in the right place at the right time. New technologies and approaches, such as high-resolution live-cell imaging and systems biology, should yield new insights into how space and time control the specificity of ubiquitin signaling.
Intense studies in the past few years on NF-κB signaling have not only uncovered the central role of ubiquitination in the regulation of IKK in different pathways, but also revealed an unexpected complexity of signaling mechanisms in these pathways. While there is strong evidence that K63 polyubiquitination is essential for IKK activation in the IL-1β pathway, the mechanism of IKK activation in the TNFα pathway appears to be more complex. Not only are Ubc13 and K63 polyubiquitination dispensable for IKK activation by TNFα, but also deletion of signaling proteins such as TRAF2, TRAF5, RIP1 or TAK1 only leads to a partial inhibition of IKK. The activated TNF receptor complex contains many proteins, including multiple ubiquitin ligases. Thus, it is possible that there is a considerable degree of redundancy in TNF signaling. For instance, K63 and other types of polyubiquitination, including linear and perhaps “mixed” polyubiquitination, may play redundant roles in IKK activation by TNFα. Unlike the IL-1 pathway in which IKK activation by TRAF6 can be reconstituted in cell-free extracts, it has not been possible to establish a cell-free system that mimics the TNFα pathway using proteins such as TRAF2, cIAP1 or HOIP. In-depth biochemical and genetic studies are required to unravel the complexity of IKK regulation by TNFα.
A common and important issue in ubiquitin research is the identification and validation of physiologically relevant ubiquitination targets. Normally, a very small fraction of a protein target is modified by ubiquitin, making it difficult to study the activity of the modified proteins and determine the modification sites. Rapid advances in mass spectrometry should help to overcome this technical hurdle. The validation of ubiquitination targets often relies on mutagenesis of the putative or confirmed ubiquitination sites. However, such mutagenesis data must be interpreted with caution, because mutations of certain residues can lead to unintended consequences, such as affecting ubiquitin binding, enzymatic functions or protein conformations. The recent finding that unanchored polyubiquitin chains have direct signaling functions in regulating the activity of TAK1, IKK and RIG-I adds another layer of complexity to ubiquitin signaling. A burning issue in NF-κB research is to sort out the role of many proposed ubiquitination targets (e.g., NEMO, TAK1, TAB2, RIPs, IRAKs, TRAFs, MALT1, BCL10, ELKS, RIG-I and TBK1) as well as unanchored polyubiquitin chains in IKK activation by different pathways. Clarifying this issue will also help to understand how DUBs such as A20 and CYLD inhibit IKK.
References
Makarova KS, Koonin EV . Archaeal ubiquitin-like proteins: functional versatility and putative ancestral involvement in tRNA modification revealed by comparative genomic analysis. Archaea 2010; pii:710303.
Finley D, Ozkaynak E, Varshavsky A . The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell 1987; 48:1035–1046.
Pickart CM . Mechanisms underlying ubiquitination. Annu Rev Biochem 2001; 70:503–533.
Hurley JH, Lee S, Prag G . Ubiquitin-binding domains. Biochem J 2006; 399:361–372.
Kirisako T, Kamei K, Murata S, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 2006; 25:4877–4887.
Adhikari A, Chen ZJ . Diversity of polyubiquitin chains. Dev Cell 2009; 16:485–486.
Johnson ES . Ubiquitin branches out. Nat Cell Biol 2002; 4:E295–298.
Chen ZJ, Sun LJ . Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 2009; 33:275–286.
Hayden MS, Ghosh S . Shared principles in NF-κB signaling. Cell 2008; 132:344–362.
Pomerantz JL, Baltimore D . Two pathways to NF-κB. Mol Cell 2002; 10:693–695.
Maniatis T . A ubiquitin ligase complex essential for the NF-κB, Wnt/Wingless, and Hedgehog signaling pathways. Genes Dev 1999; 13:505–510.
Lin L, Ghosh S . A glycine-rich region in NF-κB p105 functions as a processing signal for the generation of the p50 subunit. Mol Cell Biol 1996; 16:2248–2254.
Piwko W, Jentsch S . Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol 2006; 13:691–697.
Chen ZJ . Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol 2005; 7:758–765.
Chen ZJ, Parent L, Maniatis T . Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 1996; 84:853–862.
Ishida T, Mizushima S, Azuma S, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 1996; 271:28745–28748.
Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV . TRAF6 is a signal transducer for interleukin-1. Nature 1996; 383:443–446.
Deng L, Wang C, Spencer E, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000; 103:351–361.
Dunne A, O'Neill LA . The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE 2003; 2003:re3.
Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 1995; 270:2008–2011.
Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346–351.
Kanayama A, Seth RB, Sun L, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 2004; 15:535–548.
Kobayashi N, Kadono Y, Naito A, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 2001; 20:1271–1280.
Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD . Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-κB activation. Mol Cell Biol 2008; 28:3538–3547.
Lamothe B, Besse A, Campos AD, et al. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I κ B kinase activation. J Biol Chem 2007; 282:4102–4112.
Lamothe B, Webster WK, Gopinathan A, et al. TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun 2007; 359:1044–1049.
Yin Q, Lin SC, Lamothe B, et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol 2009; 16:658–666.
Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y . TRAF6 autoubiquitination-independent activation of the NFκB and MAPK pathways in response to IL-1 and RANKL. PLoS One 2008; 3:e4064.
Yamazaki K, Gohda J, Kanayama A, et al. Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci Signal 2009; 2:ra66.
Yamamoto M, Okamoto T, Takeda K, et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 2006; 7:962–970.
Yamamoto M, Sato S, Saitoh T, et al. Cutting edge: pivotal function of Ubc13 in thymocyte TCR signaling. J Immunol 2006; 177:7520–7524.
Xu M, Skaug B, Zeng W, Chen ZJ . A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFα and IL-1β. Mol Cell 2009; 36:302–314.
Sayama K, Yamamoto M, Shirakata Y, et al. E2 polyubiquitin-conjugating enzyme Ubc13 in keratinocytes is essential for epidermal integrity. J Biol Chem 2010; 285:30042–30049.
Trompouki E, Hatzivassiliou E, Tsichritzis T, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003; 424:793–796.
Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ . The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 2004; 14:289–301.
Windheim M, Stafford M, Peggie M, Cohen P . Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IκBα kinase. Mol Cell Biol 2008; 28:1783–1791.
Xia ZP, Sun L, Chen X, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009; 461:114–119.
Israel A . NF-κB activation: nondegradative ubiquitination implicates NEMO. Trends Immunol 2006; 27:395–397.
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ . Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22:245–257.
Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD . Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation [corrected]. Nat Cell Biol 2006; 8:398–406.
Bloor S, Ryzhakov G, Wagner S, et al. Signal processing by its coil zipper domain activates IKK gamma. Proc Natl Acad Sci USA 2008; 105:1279–1284.
Cordier F, Grubisha O, Traincard F, et al. The zinc finger of NEMO is a functional ubiquitin-binding domain. J Biol Chem 2009; 284:2902–2907.
Rahighi S, Ikeda F, Kawasaki M, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009; 136:1098–1109.
Lo YC, Lin SC, Rospigliosi CC, et al. Structural basis for recognition of diubiquitins by NEMO. Mol Cell 2009; 33:602–615.
Laplantine E, Fontan E, Chiaravalli J, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J 2009; 28:2885–2895.
Gautheron J, Courtois G . “Without Ub I am nothing”: NEMO as a multifunctional player in ubiquitin-mediated control of NF-κB activation. Cell Mol Life Sci; 67:3101–3113.
Tang ED, Wang CY, Xiong Y, Guan KL . A role for NF-κB essential modifier/IκB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IκB kinase complex by tumor necrosis factor-α. J Biol Chem 2003; 278:37297–37305.
Tokunaga F, Sakata S, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 2009; 11:123–132.
Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S . Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 2003; 115:565–576.
Zhou H, Wertz I, O'Rourke K, et al. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 2004; 427:167–171.
Abbott DW, Wilkins A, Asara JM, Cantley LC . The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 2004; 14:2217–2227.
Abbott DW, Yang Y, Hutti JE, et al. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol 2007; 27:6012–6025.
Hinz M, Stilmann M, Arslan SC, et al. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol Cell 2010; 40:63–74.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R . Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003; 424:797–801.
Kovalenko A, Chable-Bessia C, Cantarella G, et al. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003; 424:801–805.
Jaattela M, Mouritzen H, Elling F, Bastholm L . A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol 1996; 156:1166–1173.
Heyninck K, Beyaert R . The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-κB activation at the level of TRAF6. FEBS Lett 1999; 442:147–150.
Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004; 430:694–699.
Shembade N, Ma A, Harhaj EW . Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010; 327:1135–1139.
Chen G, Goeddel DV . TNF-R1 signaling: a beautiful pathway. Science 2002; 296:1634–1635.
Tartaglia LA, Goeddel DV . Two TNF receptors. Immunol Today 1992; 13:151–153.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114:181–190.
Mahoney DJ, Cheung HH, Mrad RL, et al. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc Natl Acad Sci USA 2008; 105:11778–11783.
Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689–700.
O'Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT . Ubiquitination of RIP1 regulates an NF-κB-independent cell-death switch in TNF signaling. Curr Biol 2007; 17:418–424.
Wang L, Du F, Wang X . TNF-α induces two distinct caspase-8 activation pathways. Cell 2008; 133:693–703.
Newton K, Matsumoto ML, Wertz IE, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 2008; 134:668–678.
Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009; 36:831–844.
Weil R, Israel A . Deciphering the pathway from the TCR to NF-κB. Cell Death Differ 2006; 13:826–833.
van Oers NS, Chen ZJ . Cell biology. Kinasing and clipping down the NF-κB trail. Science 2005; 308:65–66.
Ni CY, Wu ZH, Florence WC, et al. Cutting edge: K63-linked polyubiquitination of NEMO modulates TLR signaling and inflammation in vivo. J Immunol 2008; 180:7107–7111.
Wu CJ, Ashwell JD . NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-κB activation. Proc Natl Acad Sci USA 2008; 105:3023–3028.
Oeckinghaus A, Wegener E, Welteke V, et al. Malt1 ubiquitination triggers NF-κB signaling upon T-cell activation. EMBO J 2007; 26:4634–4645.
King CG, Kobayashi T, Cejas PJ, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med 2006; 12:1088–1092.
Qiao G, Li Z, Molinero L, et al. T-cell receptor-induced NF-κB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol 2008; 28:2470–2480.
Kojo S, Elly C, Harada Y, et al. Mechanisms of NKT cell anergy induction involve Cbl-b-promoted monoubiquitination of CARMA1. Proc Natl Acad Sci USA 2009; 106:17847–17851.
Zhang J, Stirling B, Temmerman ST, et al. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest 2006; 116:3042–3049.
Stilo R, Varricchio E, Liguoro D, Leonardi A, Vito P . A20 is a negative regulator of BCL10- and CARMA3-mediated activation of NF-κB. J Cell Sci 2008; 121:1165–1171.
Duwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving Malt1 ubiquitin chains. J Immunol 2009; 182:7718–7728.
Rebeaud F, Hailfinger S, Posevitz-Fejfar A, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 2008; 9:272–281.
Coornaert B, Baens M, Heyninck K, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat Immunol 2008; 9:263–271.
Hailfinger S, Lenz G, Ngo V, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2009; 106:19946–19951.
Ferch U, Kloo B, Gewies A, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2009; 206:2313–2320.
Sun L, Liu S, Chen ZJ . SnapShot: pathways of antiviral innate immunity. Cell 2010; 140:436–436 e432.
Rehwinkel J, Reis e Sousa C . RIGorous detection: exposing virus through RNA sensing. Science; 327:284–286.
Yoneyama M, Fujita T . Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008; 29:178–181.
Hornung V, Ellegast J, Kim S, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006; 314:994–997.
Pichlmair A, Schulz O, Tan CP, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006; 314:997–1001.
Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101–105.
Murali A, Li X, Ranjith-Kumar CT, et al. Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J Biol Chem 2008; 283:15825–15833.
Satoh T, Kato H, Kumagai Y, et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA 2010; 107:1512–1517.
Seth RB, Sun L, Ea CK, Chen ZJ . Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005; 122:669–682.
Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6:981–988.
Xu LG, Wang YY, Han KJ, et al. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell 2005; 19:727–740.
Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167–1172.
Gack MU, Shin YC, Joo CH, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007; 446:916–920.
Zeng W, Sun L, Jiang X, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010; 141:315–330.
Zeng W, Xu M, Liu S, Sun L, Chen ZJ . Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell 2009; 36:315–325.
Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006; 439:208–211.
Zhao T, Yang L, Sun Q, et al. The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat Immunol 2007; 8:592–600.
Lin R, Yang L, Nakhaei P, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem 2006; 281:2095–2103.
Friedman CS, O'Donnell MA, Legarda-Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep 2008; 9:930–936.
Zhang M, Wu X, Lee AJ, et al. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem 2008; 283:18621–18626.
Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science 2007; 318:1628–1632.
Ting JP, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity 2008; 28:285–287.
Kanneganti TD, Lamkanfi M, Nunez G . Intracellular NOD-like receptors in host defense and disease. Immunity 2007; 27:549–559.
Inohara N, Koseki T, del Peso L, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J Biol Chem 1999; 274:14560–14567.
Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem 2001; 276:4812–4818.
Franchi L, Warner N, Viani K, Nunez G . Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev 2009; 227:106–128.
Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J 2008; 27:373–383.
Yang Y, Yin C, Pandey A, et al. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem 2007; 282:36223–36229.
Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 2009; 106:14524–14529.
Bertrand MJ, Doiron K, Labbe K, et al. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009; 30:789–801.
Hitotsumatsu O, Ahmad RC, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008; 28:381–390.
Wu ZH, Miyamoto S . Many faces of NF-kappaB signaling induced by genotoxic stress. J Mol Med 2007; 85:1187–1202.
Mabb AM, Wuerzberger-Davis SM, Miyamoto S . PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat Cell Biol 2006; 8:986–993.
McCool K, Miyamoto S . A PAR-SUMOnious mechanism of NEMO activation. Mol Cell 2009; 36:349–350.
Stilmann M, Hinz M, Arslan SC, et al. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IκB kinase activation. Mol Cell 2009; 36:365–378.
Wu ZH, Shi Y, Tibbetts RS, Miyamoto S . Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science 2006; 311:1141–1146.
Wu ZH, Wong ET, Shi Y, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell 2010; 40:75–86.
Grassmann R, Aboud M, Jeang KT . Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 2005; 24:5976–5985.
Shembade N, Harhaj NS, Yamamoto M, Akira S, Harhaj EW . The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-κB activation. J Virol 2007; 81:13735–13742.
Yu Q, Minoda Y, Yoshida R, et al. HTLV-1 Tax-mediated TAK1 activation involves TAB2 adapter protein. Biochem Biophys Res Commun 2008; 365:189–194.
Wu X, Sun SC . Retroviral oncoprotein Tax deregulates NF-κB by activating Tak1 and mediating the physical association of Tak1-IKK. EMBO Rep 2007; 8:510–515.
Shembade N, Harhaj NS, Liebl DJ, Harhaj EW . Essential role for TAX1BP1 in the termination of TNF-α-, IL-1- and LPS-mediated NF-κB and JNK signaling. EMBO J 2007; 26:3910–3922.
Shembade N, Harhaj NS, Parvatiyar K, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol 2008; 9:254–262.
Luftig M, Yasui T, Soni V, et al. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK α-dependent noncanonical NF-κB activation. Proc Natl Acad Sci USA 2004; 101:141–146.
Luftig M, Prinarakis E, Yasui T, et al. Epstein-Barr virus latent membrane protein 1 activation of NF-κB through IRAK1 and TRAF6. Proc Natl Acad Sci USA 2003; 100:15595–15600.
Chung YH, Jhun BH, Ryu SC, et al. STP-C, an oncoprotein of herpesvirus saimiri augments the activation of NF-κB through ubiquitination of TRAF6. J Biochem Mol Biol 2007; 40:341–348.
An J, Mo D, Liu H, et al. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-κB activation. Cancer Cell 2008; 14:394–407.
Xiao G, Harhaj EW, Sun SC . NF-kappaB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell 2001; 7:401–409.
Liao G, Zhang M, Harhaj EW, Sun SC . Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 2004; 279:26243–26250.
Sun SC . Controlling the fate of NIK: a central stage in noncanonical NF-κB signaling. Sci Signal; 3:pe18.
Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 2007; 131:682–693.
Vallabhapurapu S, Matsuzawa A, Zhang W, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol 2008; 9:1364–1370.
Zarnegar BJ, Wang Y, Mahoney DJ, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9:1371–1378.
Elsasser S, Finley D . Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 2005; 7:742–749.
Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009; 137:133–145.
Hofmann RM, Pickart CM . In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem 2001; 276:27936–27943.
Jacobson AD, Zhang NY, Xu P, et al. The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 S proteasome. J Biol Chem 2009; 284:35485–35494.
Acknowledgements
We thank Xin Cai and Brian Skaug for critically reading the manuscript. Research in our laboratory is supported in part by grants from NIH (RO1-GM63692) and the Welch Foundation (I-1389).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is cited by
-
Exosomal USP13 derived from microvascular endothelial cells regulates immune microenvironment and improves functional recovery after spinal cord injury by stabilizing IκBα
Cell & Bioscience (2023)
-
TRIM67 alleviates cerebral ischemia‒reperfusion injury by protecting neurons and inhibiting neuroinflammation via targeting IκBα for K63-linked polyubiquitination
Cell & Bioscience (2023)
-
Simultaneous reduction of all ORMDL proteins decreases the threshold of mast cell activation
Scientific Reports (2023)
-
TRIM22 activates NF-κB signaling in glioblastoma by accelerating the degradation of IκBα
Cell Death & Differentiation (2021)
-
The role of ubiquitination in tumorigenesis and targeted drug discovery
Signal Transduction and Targeted Therapy (2020)
