
Abstract
Human rhodopsin kinase (RK) and a carboxyl terminus-truncated mutant RK lacking the last 59 amino acids (RKC) were expressed in human embryonic kidney 293 cells to investigate the role of the carboxyl terminus of RK in recognition and phosphorylation of rhodopsin. RKC, like the wild-type RK, was detected in both plasma membranes and cytosolic fractions. The Cterminal truncated rhodopsin kinase was unable to phosphorylate photo-activated rhodopsin, but possesses kinase activity similar to the wild-type RK in phosphorylation of small peptide substrate. It suggests that the truncation did not disturb the gross structures of RK catalytic domain. Our results also show that RKC failed to translocate to photo-activated rod out segments. Taken together, our study demonstrate the carboxyl terminus of RK is required for phosphorylation of photo-activated rhodopsin and strongly indicate that carboxyl-terminus of RK may be involved in interaction with photo-activated rhodopsin.
Similar content being viewed by others
Introduction
Rhodopsin kinase (RK) is a member of G protein-coupled receptor kinase (GPRK) family and it specifically phosphorylates photo-activated rhodopsin (Rho*) in retinal photoreceptor cells. The phosphorylation of Rho* prevents activation of G protein transducin, promotes binding of the regulatory protein arrestin to phosphorylated Rho*, and thus terminates the activation pathway of visual transduction1, 2. This process is often called desensitization of photo-receptor, which physiologically relates to dark-adaptation. Genetic mutations of RK gene, which leads to loss of RK function and have recently been implicated in human Oguchi disease, a recessively inherited form of stationary night blindness3.
RK contains three functional domains: amino-terminal domain, catalytic domain, and carboxyl-terminal domain4. Carboxyl terminus of RK consists of a conserved region conserved in all GPRKs members and a variable region with the unique farne-sylation site which differs from other GPRKs. Mutations of RK at the farnesylation or autophosphorylation sites greatly impair its ability to phosphorylate Rho* and it is therefore generally accepted that carboxyl terminus of RK is involved in regulatory functions5, 6. Mutations occurring in the variable region of RK carboxyl terminus and catalytic domain cause Oguchi disease in human3. This study further demonstrated that deletion of the variable region of RK carboxyl terminus abolished its ability to phosphorylate Rho*.
Materials and Methods
Construction of pcDNA3-RK and pcDNA3-RKC
Human RK cDNAs were cloned by screening a human retinoblastoma cDNA library (Clontech) using bovine RK cDNA as the probe. A full length of RK cDNA clone was constructed by ligation of three clones containing partial RK cDNA utilizing the Kpn I and EcoR I sites within RK. RKC lacking the 59 amino acid residues (505-563) from the C-terminus was obtained by digestion of RK cDNA with EcoR I utilizing the EcoR I site 177 bp upstream of the stop codon. The resultant RK and RKC cDNAs were confirmed by sequencing, subcloned into pcDNA3 (Invitrogen), and designated as pcDNA3-RK and pcDNA3-RKC, respectively.
Preparation of anti-RK polyclonal antibodiesy
Human RK cDNA was subcloned into pPET-3a and RK was overexpressed in E. coli. The inclusion bodies were washed with 2 M urea in 100 mM Tris-HC1, pH 8.0, 1% Triton X-100 solution, dissolved in 6M guanidine hydrochloride, and dialyzed in 0.1 M Tris-HC1, pH 8.0. The purity of recombinant RK obtained was over near 80 % by commassie blue staining following SDS-PAG. Antisera were raised in Balb/c mice using total 50 μg RK with complete Freund's adjuvant.
Expression of RK and RKC in human embryonic kidney (HEK) 293 cells
HEK 293 cells were cultured in MEM (GIBCO BRL) with 10 % fetal bovine serum (GIBCO BRL), 100 μg/ml streptomycin, and 100 units/ml penicillin. Forty eight hours after transiently transfected with pcDNA3-RK or pcDNA3-RKC by calcium phosphate co-precipitation, the 293 cells were harvested and cytosolic fractions and plasma membranes were prepared as described7. Expression of RK and RKC were examined by Western blot analysis using the anti-RK polyclonal antibodies raised and visualized by Enhanced Chemi-Luminescence kit (ECL, Amersham, Buckinghamshire, UK).
Rhodopsin phosphorylation assay
Bovine rod outer segments (ROS) were prepared from fresh retina and washed with 4M urea as described8, 9. Rhodopsin was the major component of ROS protein (> 95 %). RK activity was measured using ROS containing 10 μM rhodopsin as the substrate in the reaction mixture of 75 mM TrisHCl, pH 7.5, 10 mM Mg2+, 5 mM DTT, 100 μM [γ-32P]ATP (500 dpm/pmol), 1 μM H-89 (cAMP-dependent kinase inhibitor, Ki = 0.048 μM), and 13 μM chelerythrtine chloride (protein kinase C inhibitor, Ki = 0.66 (M). The cytosolic fractions of the transfected HEK 293 cells with comparable amount of RK and RKC were used in the phosphorylation assay. The phosphorylation reaction was initiated by addition of [γ-32P]ATP and ROS, incubated at 30 °C under the illumination of white light, and stopped by addition of the SDS-PAGE sample loading buffer. Recombinant bovine GRK2 purified from E. coli (generously provided by Dr. Lin Li at Shanghai Institute of Biochemistry) was used as a positive control. All samples were resolved on 10 % polyacrylamide gels and phosphorylation of Rho* was visualized by autoradiography.
Peptide phosphorylation assay
The kinase activity of RK and RKC towards the peptide substrate of RRREEEEESAAA, which has been used to measure activity of RK and GPRK210, 11 was determined using phosphocellulose method. The cytosolic fraction of the transfected HEK 293 was used in the phosphorylation assay as a source of rhodopsin kinase. The reaction mixture (20 μl) contained 1 mM peptide substrate in buffer of 20 mM TrisHCl, pH 7.5, 7.5 mM Mg2+, 2 mM EDTA, 1 mM DTT, 100 μM [γ-32P]ATP (300-500 dpm/pmol), 1 μM H-89 and 13 μM chelerythtine chloride. The kinase reaction was initiated by addition of [γ-32P]ATP and the peptide, incubated at 30 °C for 20 min, and terminated by addition of trichloroaceticacid to 10 % final concentration. The samples were centrifuged at 13,000 x g for 15 min, and the supernatants were spotted in duplicate onto P81 phosphocellulose filter (Whatman). The filters were washed in 75 mM phosphoric acid, and the radioactivity remained on the filters was determined in a scintillation counter. The control experiments for nonspecific phosphorylation were conducted in the absence of the peptide substrate.
RK translocation assay
Translocation of RK or RKC to photo-activated ROS was determined as described12. Briefly, an aliquot of the cytosolic fractions of the transfected HEK 293 cells was incubated with ROS membranes (containing 10 μM rhodopsin) in translocation buffer of 50 mM Na2HPO4, pH 7.4, 100 mM NaCl, 1 mM Mg2+ 5 mM DTT, 1 μM H-89, 13 μM chelerythtine chloride, and a cocktail of protease inhibitors. The incubation was performed at 25 °C for 4 min in dark or under the illumination of white light. The samples were then centrifuged at 300,000 g for 15 min at 4 °C. The supernatant fractions were quickly collected and pelleted membranes were resuspended in translocation buffer. Both fractions were subjected to SDS-PAGE and immunoblotted by the RK antibodies.
Results
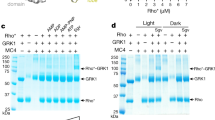
Mammalian expression system (HEK 293 cells) was chosen to express RK and RKC to avoid any potential defect in post-translational modification. Expression of RK and RKC in HEK 293 cells was examined by Western Blot analysis using anti RK antibodies. Western results show human rhodopsin kinase as a single major band of ∼ 63 kDa and present in both the cytosolic fractions and plasma membranes in pcDNA3-RK transfected HEK 293 cells. The apparent molecular mass Yu QM et al. of the expressed human RK consistents with previously reported size for its bovine counterpart5. The C-terminal truncated rhodopsin kinase (∼ 50 kDa) migrated faster than the wild-type RK on gel (Fig 1). Expression level of RKC was comparable to that of wild-type RK and the distribution of RKC in cytosolic fraction and on plasma membranes was not altered by the carboxyl terminal truncation significantly under the conditions of overexpression used in this study (Fig 1). These results indicate that the truncation at C-terminus of RK did not affect its expression and membrane association.

Distribution of RK and RKC in the cytosolic and membrane fractions in transfected HEK 293 cells. The cytosolic (lane 1-3) and plasma membrane fractions (lane 4-6) were prepared from cells transfected with pcDNA3 (lane 1, 4), pcDNA3-RK (lane 2, 5), or pcDNA3-RKC (lane 3, 6). and Western analysis was done using RK-specific antibodies.
The cytosolic fractions of transfected HEK 293 cells containing comparable amounts of wild-type and the truncated RK were analyzed for the specific kinase activity against photo-activated rhodopsin. As shown in Fig 2, expression of wild-type RK caused phosphorylation of Rho*. Phosphorylation of activated rhodopsin by human RK was time-dependent. Rho* phosphorylation was detected in 1 min and the reaction approached maximum in 9 min. However, under the same experimental conditions, the expression of the truncated rhodopsin kinase RKC had little, if any, effect on phosphorylation of the same substrate. The extent of Rho* phosphrylation in samples containing RKC increased slightly as reaction time increased, but was not significantly different from control reaction containing cytosolic extract from cells transfected with vector pcDNA3 even after 9 min incubation (Fig 2, lanes 2 and 9). The results clearly demonstrated that truncation at the C-terminus of rhodopsin kinase greatly impaired its ability to phosphorylate Rho* and suggest that the 59 residues at the C-terminus of rhodopsin kinase are essential for phosphorylation of its natural substrate, photo-activated rhodopsin.

Phosphorylation of Rho* by RK and RKC. Rhodopsin kinase activities in the cytosolic fractions from cells transfected with pcDNA3 (lane 1), pcDNA3-RK (lane 2-5), or pcDNA3-RKC (6-9) were determined using Rho* as a substrate. The extents of Rho* phosphorylation were examined following gel electrophoresis and radioautography. Equivalent amounts of RK and RKC, as verified by Western analysis, were applied in the Rho* phosphorylation assay and the reactions were incubated for 0 (lane 2, 6), 1 (lane 3, 7), 3 (lane 4, 8), and 9 (lane 1, 5, 9, 10) min. Lane 10, a positive control containing 200 ng purified GRK2. The blot shown is a representative autoradiography of three experiments.
The synthetic peptide RRREEEEESAAA has been used as a substrate of RK and GPRK2 in studies of their functions10, 11. To investigate whether the carboxyl terminal truncation of RK affects gross conformation of RK and in turn impair the function of its catalytic domain, the kinase activity of RK and RKC towards the small peptide substrate was compared. As presented in Fig 3, RKC was able to phosphorylate the small peptide substrate, as RK does, and its kinase activity against the peptide substrate was comparable to that of wild-type RK. This result

Phosphorylation of peptide substrate by RK and RKC. Rhodopsin kinase activities in the cytosolic fractions from cells transfected with pcDNA3, pcDNA3-RK, or pcDNA3-RKC were determined using RK substrate peptide RRREEEEESAAA. The extents of phosphorylation of the peptide substrate were determined by [32P] incorporation into the substrate. Equivalent amounts of RK and RKC, as verified by Western analysis, were applied in the assay, Relative kinase activity was calculated against the control sample (trausfection of vector pcDNA3), and the results were mean ± S.E. of three independent experiments.
One of the explanations for incompetence of RKC to phosphorylate Rho* could be that the truncation greatly affected its ability to interact with rhodopsin. To test this, the capabilities of the wild-type and truncated rhodopsin kinase to bind Rho* on bovine ROS was assessed. Rhodopsin takes up over 80 % of the surface in ROS13 and RK binds to rhodopsin on the membranes and catalyzes its phosphorylation13. As shown in Fig 4, without illumination, association of rhodopsin kinase to ROS membrane was not detected. Upon photo-activation, the wild type kinase transferred to ROS membranes (Fig 4). In strong contrast, however, RKC was unable to translocate to photo-activated ROS membranes (Fig 4). These data strongly indicate that truncation at the C-terminus of RK may affect its interaction with photo-activated rhodopsin.

Translocation of RK and RKC to ROS. Translocation assays were carried out in dark (D) or under the luminescence of white light (L) for 4 min in the presence of the cytosolic fractions from HEK 293 cells transfected with pcDNA3 (control), pcDNA3-RK (RK) , or pcDNA3-RKC (RKC). At end of the reaction, reaction mixtures were centrifuged. The supernatants (S) and the pelleted ROS membranes (M) were subjected to SDS-PAGE and then immunoblotted by the antihuman RK antibodies.
Discussion
As a member of GPRK family, RK is homologous to other GPRKs at the amino terminal and the central region (catalytic domain)6. Major differences in primary sequence of RK from other GPRKs are located at its carboxyl terminus, which uniquely undergoes post-translational modifications of farnesylation, proteolysis and α -carboxyl methylation5. These modifications appear to be important for the light-dependent association of the kinase with Rho*, a processes usually called translocation, and consequently for the kinase activity to phosphorylate Rho*12, 14. Though the ability of RK to translocate to photo-activated ROS membranes was abolished by the point mutation of the farnesylation site at its carboxyl terminus, approximately 25 % of its activity against Rho* still remains5. Our current study show that further deletion of amino acid 505-563 on the C-terminus of RK not only abolished its light-dependent translocation but also totally demolished its ability to phosphorylate Rho*. The results indicate that the C-terminal structure of RK is essential for phosphorlation of activated rhodopsin and the C-terminal structure of RK, in addition to hosting the post-translational modifications, may also be involved in regulation of its activity towards its physiological substrate.
Mutagenesis has been widely used in the studies of structure and function of enzymes including kinase and phosphotase, but one major concern that the mutation may disrupt the overall conformation of enzymes remains. In attempt to address this question, a small peptide substrate was applied to test the relative enzymatic activity of RKC versus RK though the peptide was a less efficient substrate (Km ≈ 2 mM) compared to rhodopsin (Km = 4 μM) for RK10. Our data showed that the kinase activity of RKC against the peptide substrate was comparable to that of the wild-type, suggesting the significant disruption of conformation of the catalytic domain by deletion of the C-terminal residues of RK was very unlikely.
Under physiological conditions, the wild-type RK appears in cytosol in the dark but translocates to and then tightly associates with ROS membranes in the light12. When overexpressed in HEK 293 cells, however, RK appeared in both the cytosolic fractions and on plasma membranes. Removal of carboxyl terminal 59 amino acids (505-563) of RK did not alter its apparent distribution, indicating the general ability to associate with cell plasma membranes remains intact in RKC. Therefore, loss of activities of RKC to translocate to photo-activated ROS membranes and to phosphorylate Rho* strongly indicates that C-terminus may be involved in interaction with photo-activated rhodopsin. There has been reported that GPRK4 (γ-form), one of the four native splicing variants of GPRK4, lacking the 47 amino acids (515-562) at the variable region of the carboxyl terminus of a-form, is unable to phosphorylate Rho*15. In addition, mutations found in human Oguchi disease also give some clues that carboxyl terminus is indeed required for RK function3. Our results in this study demonstrate that the carboxyl terminus of RK is required for phosphorylation of photo-activated rhodopsin and strongly indicate that C-terminus may be involved in interaction with photo-activated rhodopsin.
Abbreviations
- Rho:
-
photo-activated rhodopsin
- RK:
-
rhodopsin kinase
- ROS:
-
rod outer segments
- SDS:
-
sodium dodecyl sulfate
- PAGE:
-
polyacrylamide gel electrophoresis
- PKA:
-
cyclic adenosine monophosphate-dependent protein kinase
- PKC:
-
calcium-dependent protein kinase
- GPRK:
-
G protein-coupled receptor kinase
References
Wilden U, Hall SW, Huhn H . Phosphodiesterase activation by photoexcitd rhodopsin is quenched when rhodopsin is phosphoprylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA 1986; 83:1174–8.
Bennett N, Sitaramayya A . Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein(arrestin). Biochemistry 1988; 27:1710–5.
Yamamoto S, Sippel KC, Berson EL Dryja TP . Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997; 15:175–8.
Inglese J, Freedman NJ, Koch JW and Lefkowitz RJ . Structure and mechanism of the G protein-coupled receptor kinase. J Biol Chem 1993; 268:23735–8.
Inglese J, Glickman JF, Lorenz W . Isoprenylation of a protein kinase: Requirement of farnesylation/alpha-carboxyl methylation for full enzymatic activation of rhodopsin kinase. J Biol Chem 1992; 267:18991–8.
Richard TP, Inglese J, Lefkowitz RJ . Protein kinase that phosphorylates activated G protein-coupled receptors. FASEB J 1995; 9:175–82.
Murga C, Esteban N, Ruiz-Gomez A, Mayor F Jr . The basal subcellular distribution of β(adrenergic receptor kinase is independent of G-protein β γ subunit. FEBS Letter 1997; 409:24–8.
Papermaster DS . Preparation of antibodies to rhodopsin and the large protein of rod outer segments. Methods Enzymol 1982; 81:48–52.
Palczewski K, McDowell JH, Hargrave PA . Purification and characterization of rhodopsin kinase. Biochemistry 1988; 27:2306–3.
Onorato JJ, Palczewski K, Regan JW, Caron MG, Lefkowitz RJ, Benovic JL . Role of acidic amino acids in peptide substrates of the beta-adrenergic receptor kinase and rhodopsin kinase. Biochemistry 1991; 30:5118–25.
Chen CY, Dion SB, Kim CH, Benovic JL . Beta-adrenergic receptor kinase Agonist-dependent receptor binding promotes kinase activation. J Biol Chem 1993; 268:7825–31.
Inglese J, Koch WJ, Caron MG . Isoprenylation in regulation of signal transduction by G protein-coupled receptor kinase. Nature 1992; 359:147–50.
Papermaster DS, Dreyer WJ . Rhodopsin content in the outer segment membrane of bovine and frog retinal rods. Biochemistry 1974; 13:2438–44.
Kuhn H . Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry 1978; 21:4389–95.
Sallese M, Mariggio S, Collodel G, Moretti E, Piomboni P, Baccetti B, DeBlasi A . G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. J Biol Chem 1997; 272:15294–8.
Acknowledgements
This research was supported by research grants from National Natural Science Foundation of China (39630130 and 39625015), Chinese Academy of Sciences (KJ951-B1-608 and KY951-A1-301), Shanghai Research Center of Life Sciences, Shanghai Educational Development Foundation, and German Max-Planck Society. The authors wish to thank Dr. Lin Li for recombinant bovine GRK2 and Jianfang Din for technical assistance help in preparing anti-RK antibodies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, Q., Cheng, Z., Zhao, J. et al. Carboxyl terminal of rhodopsin kinase is required for the phosphorylation of photo-activated rhodopsin. Cell Res 8, 303–310 (1998). https://doi.org/10.1038/cr.1998.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.1998.30
