
Abstract
Interleukin-17 (IL-17), a member of the IL-17 cytokine family, plays a crucial role in mediating the immune response against extracellular bacteria and fungi in the lung. Although there is increasing evidence that IL-17 is involved in protective immunity against H1 and H3 influenza virus infections, little is known about the role of IL-17 in the highly pathogenic H5N1 influenza virus infection. In this study, we show that H5N1-infected IL-17 knockout (KO) mice exhibit markedly increased weight loss, more pronounced lung immunopathology and significantly reduced survival rates as compared with infected wild-type controls. Moreover, the frequency of B cells in the lung were substantially decreased in IL-17 KO mice after virus infection, which correlated with reduced CXCR5 expression in B cells and decreased CXCL13 production in the lung tissue of IL-17 KO mice. Consistent with this observation, B cells from IL-17 KO mice exhibited a significant reduction in chemokine-mediated migration in culture. Taken together, these findings demonstrate a critical role for IL-17 in mediating the recruitment of B cells to the site of pulmonary influenza virus infection in mice.
Similar content being viewed by others


Introduction
Influenza virus infections have caused substantial human morbidity and mortality over the past centuries.1, 2 In recent years, novel subtypes of influenza viruses, such as the highly pathogenic H5N1 and the pandemic H1N1, have been shown to be transmitted directly from animals to the human population.3, 4 Despite the availability of vaccines and antiviral therapies, H5N1 influenza virus infections still cause considerable severe disease with a high death rate. Mouse and human H5N1-mediated respiratory disease can be fatal and exhibits clinical features of dysregulated virus-induced cytokine production, extensive inflammatory infiltration and severe histopathological pulmonary injury.1, 5 Following H5N1 virus infection, systemic and exaggerated release of pro-inflammatory cytokines by rapidly proliferating immune cells and airway epithelial cells results in a cytokine storm and pulmonary damage. A virus-induced cytokine storm has been shown to be highly lethal among young healthy adults, probably due to the strong immune response in these subjects.6 Although extensive investigations have centered around studying viral genetics, ecology and geography of viral transmission, and clinical diagnosis and management of H5N1 infection, the dysregulated host immune response is much less defined.5
Interleukin-17 (IL-17), produced by cells of both the adaptive and innate immune systems, functions as a potent pro-inflammatory cytokine and plays a crucial role in both chronic autoimmune pathogenesis7 and host defenses against diverse pulmonary pathogens.8 Previous studies have demonstrated that IL-17 plays a beneficial role in infections by various pathogens, including pulmonary Mycobacterium bovis, Klebsiella pneumonia and vaccinia virus.9, 10, 11 The involvement of IL-17 in protection against influenza virus infection has also been indicated. Studies in vivo have demonstrated that IL-17 can protect mice against lethal infection with both A/Puerto Rico/8/34 (H1N1) and A/Alaska/6/77 (H3N2) influenza virus, which is accompanied by an enhanced early influx of neutrophils into the lung.12 Other studies using IL-10 knockout (KO) mice have also demonstrated that IL-17 can protect naive mice against a lethal dose of A/Puerto Rico/8/34 via unique mechanisms other than established Th1-mediated protection.13 In a study of prophylactic treatment with bacterially derived immunomodulators, the Th17 inflammatory response directly contributed to the enhanced survival of mice following A/Puerto Rico/8/34 challenge.14 However, previous studies on IL-17 transgenic mice and IL-17 receptor KO mice also reported that IL-17 may act as a ‘double-edged sword’ and contributes to pulmonary immunopathology.15, 16 Elevated IL-17 serum levels have also been found in patients with severely pandemic H1N1 influenza infections, which may potentiate lung inflammation and its fatal consequence.17 All of these studies highlight the need for further research to clarify the role of IL-17 during influenza virus infection, especially during highly pathogenic H5N1 virus infection.
Accumulating evidence has defined an important role for B cells in virus clearance and host survival during primary influenza virus infections.18 The protective role of B cells appears to be mediated, at least in part, through antibody production both in the early phase of virus control mediated by IgM and during later stages via neutralizing isotype-switched, virus-specific antibodies that are required for optimal virus clearance and protection from virus-induced pathology.19, 20, 21 Although the mechanisms involved in the induction and modulation of the B-cell response have been extensively investigated, whether and to what extent IL-17, a relatively new pro-inflammatory cytokine, modulates the B-cell response during influenza virus infection remains largely unclear. As the IL-17 receptor is reportedly expressed on murine B cells,22, 23, 24 the potential role of IL-17 signaling in B-cell responses needs to be further investigated. In this study, we used IL-17 KO mice to determine the function of IL-17 in regulating B-cell functions during H5N1 influenza virus infection in mice.
Materials and methods
Immunostaining
For multicolor flow cytometric analysis, cell samples were stained with the following monoclonal antibodies specific for phenotypic markers: anti-B220-FITC (clone RA3-6B2), anti-B220-PE (clone RA3-6B2), anti-Gr1-FITC (clone RB6-8C5), anti-CD11c-FITC (clone N418), anti-CD3-PE (clone 145-2C11), anti-NK1.1-PE (clone PK136) and anti-CD11b-PE (clone M1/70) from Biolegend (San Diego, CA, USA); anti-γσTCR-FITC (clone GL3), anti-CXCR5-FITC (clone 2G8) and its isotype-matched control antibody from BD Biosciences Pharmingen (San Diego, CA, USA). For immunohistochemical analysis, sections were stained with the following antibodies or reagents: anti-mouse CD45R/B220-biotin (clone RA3-6B2) from Biolegend, rabbit polyclonal antibody to BCA1 and its isotype-matched control antibody from Abcam (Cambridge, UK), polyclonal goat anti-rabbit immunoglobulin–biotin from DAKO (Glostrup, Denmark), streptavidin–HRP from Cell Signaling Technology (Beverly, MA, USA) and a DBA substrate kit from Zymed Laboratories, Inc. (San Francisco, CA, USA).
Virus preparation
Influenza type A virus, A/Vietnam/1194/2004 (H5N1) (VN1194), was propagated in the allantoic cavity of 10-day-old embryonated hens' eggs at 37 °C for 27 h, as previously described.5 Briefly, allantoic fluid was collected and stored in aliquots at −70 °C. For inactivated virus preparation, the allantoic fluid was concentrated and purified in a 10–50% sucrose gradient by centrifugation at 25 000 g, 4 °C for 2 h. The purified virus was inactivated with 0.25% formalin (v/v) at 4 °C for 7 days. The inactivated virus was further purified with Amicon Ultra Membrane (4208) (Millipore, Billerica, MA, USA), with a molecular weight cutoff at 30 kDa. The purified virus was resuspended in phosphate-buffered saline (PBS). Inactivation of the virus was confirmed by the absence of cytopathic effects and detectable hemagglutination in the supernatant of two consecutive tissue culture infectious dose-50 assays by the method of Reed and Muench.25
Animal models and viral challenge
IL-17 KO mice on a C57BL/6 background and wild-type control C57BL/6 female and male mice were 6–8 weeks old at the time of challenge. IL-17 KO mice were obtained from Dr Yoichiro Iwakura (Institute of Medical Science, The University of Tokyo, Tokyo, Japan). C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were maintained in a specific pathogen-free animal facility at The University of Hong Kong. Mice were kept in biosafety level-3 housing and given access to standard food and water. All experimental protocols followed the standard operating procedures of the approved biosafety level-3 animal facilities and were approved by the Institutional Animal Ethics Committee. The 50% lethal dose (LD50) of VN1194 was determined in mice after serial dilution of the stock, and 10LD50 were used for viral challenges. Mice were lightly anesthetized with isoflurane and were challenged intranasally with 20 µl infectious virus diluted in PBS. Following infection, mice were monitored daily for weight loss, signs of clinical illness and survival. Mice were killed at the indicated time points. The remaining mice in each group were observed daily for weight loss and survival for 14 days.
Lung fixation and histopathological assessment
Mice were killed at the indicated time points, and lungs were inflated with 10% neutral buffered formalin for at least 24 h before processing and embedding. Lung tissue was sectioned at 5-µm thickness and stained with hematoxylin and eosin for histopathological evaluation. Slides were examined in a blinded manner and scored with a semiquantitative system according to the relative degree of inflammation and tissue damage.16, 26, 27 The cumulative scores of inflammatory infiltration, degeneration and necrosis provided the total score per animal, and the average score of mice in each group was taken as the total score for that group. Lung infiltration of inflammatory cells was scored as follows: 0, no inflammation; 1, mild peribronchial and peribronchiolar infiltrates, extending throughout <10% of the lung; 2, moderate inflammation covering 10–50% of the lung; 3, severe inflammation involving over one-half of the lung. Degeneration of bronchi and bronchiolar epithelium, alveoli degeneration and collapse was scored as follows: 0, no degeneration; 1, little vacuolar degeneration of bronchi and bronchial epithelium cells, normal pulmonary alveoli; 2, mild necrosis of bronchi and bronchial epithelium, mild alveoli damage; 3, severe degeneration. Necrosis was scored as follows: 0, no necrosis; 1, mild necrosis with scant exudate; 2, marked necrosis with abundant exudate; 3, severe interstitial edema around blood vessels, apparent injured parenchyma and degenerated alveolar epithelial cells with greater infiltration of inflammatory cells. The alveolar architecture is obliterated due to thickening of the alveolar septa and flooding of the alveolar lumina with alveolar macrophages, neutrophils and erythrocytes, combined with fibrin, edema fluid and cellular debris.
RNA extraction and quantitative PCR analysis with reverse transcriptase
Mice were euthanized at the indicated time points. Lungs were homogenized in PBS using an electric tissue grinder for RNA purification. Total RNA samples from lung homogenates were prepared with an RNeasy Kit (Qiagen, Hilden, Germany) and reverse transcribed with SuperScript III First-Strand Synthesis SuperMix (Invitrogen, Carlsbad, CA, USA). Real-time PCR was performed using Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) according to the manufacturer's instructions with an Applied Biosystems Prism 7900HT real-time PCR system (Foster City, CA, USA). Real-time PCR reactions were set up under the following conditions: 95 °C for 2 min, 40 cycles of 95 °C for 15 s and 60 °C for 30 s. The threshold cycle (CT) of gene products was determined and set to the log-linear range of the amplification curve and kept constant. Relative expression of CXCL13 was calculated as with normalization to the corresponding β-actin values. The copy number of the nucleoprotein (NP) gene was calculated using an NP (GenBank record AY651498, 1485 bp)-containing plasmid of known concentration as a standard. The sequences of primers designed to span one intron each for the CXCL13 and β-actin genes were as follows: CXCL13 forward, 5′-GATCGGATTCAAGTTACGCC-3′ and reverse 5′-CAGGCAGCTCTTCTCTTACTCA-3′; β-actin forward, 5′-CCACACCCGCCACCAGTTCGC-3′ and reverse 5′-CTTTGCACATGCCGGAGCCGTT-3′; NP forward, 5′- GAACCAGAAGCGGAGGAAACA-3′ and reverse 5′-CTGCCATAATGGTCGCTCTT -3′.
Isolation of B cells
Spleens from mice were harvested from IL-17 KO or C57BL/6 mice for B-cell isolations. Splenic cell suspensions were prepared and red blood cells depleted with ACK buffer and subsequent washing. Cells were then labeled with CD45R (B220) MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Splenic B cells were isolated by magnetic-activated cell sorting enrichment using an LS column (Miltenyi Biotec). The purity of collected cells was examined with an EPICS-Altra flow cytometer (Beckman Coulter, Fullerton, CA, USA) and was routinely greater than 96%. Purified splenic B cells were then subjected to a CXCL13 chemotaxis assay.
B-cell migration assay
B-cell chemotaxis assays were conducted in 24-well plates containing transwell inserts with a 5-µm pore size (3421) (Corning Costar, Cambridge, MA, USA). Purified B cells (2×106 cells/ml) were resuspended in RPMI media with 10% fetal calf serum (R10). Migration medium (0.6 ml) containing 400 ng/ml CXCL13 (Peprotech, Rocky Hill, NJ, USA) or control medium R10 were added to the lower chamber. Media with B cells contained in suspension (0.1 ml) was added to the upper chamber and the transwell plate was incubated at 37 °C for 5 h. Cells in the lower and upper chambers were then collected, diluted to an equal volume of 0.6 ml, and counted with a flow cytometer (Beckman Coulter) under a constant medium flow rate. Each treatment was performed with triplicate transwells. The percentage of chemotaxis was calculated as follows: %migration=migrated cells×100/input cells, %B-cell migration=%chemokine migration−%control migration.
Inactivated virus stimulation
To assess the immune response induced by inactivated virus in vivo, wild-type and IL-17 KO mice received two doses (10 µg/dose) of inactivated virus in 50 µl PBS on alternating days. Mice were killed 24 h after the second administration. Total lung cells were isolated as described.28 Briefly, mice were killed and perfused with PBS via injection into the right ventricle, which flushes blood vessels in the lungs. Lung tissue was collected for type II collagenase and DNase I (bovine pancreas) (Merck, Whitehouse Station, NJ, USA) digestion. After treatment with ACK buffer, the total number of cells was counted. Frequencies of various immune cell populations in the lung were determined by immunostaining and flow cytometric analysis.
Statistical analysis
Data are expressed as mean±standard deviation. Statistical comparisons were calculated by the Student's t-test. For the survival curve of virus-infected mice, the Kaplan–Meier analysis was used. A value of P<0.05 was considered statistically significant.
Results
IL-17 KO mice infected with H5N1 influenza virus display higher mortality
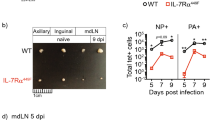
To examine the role of IL-17 during H5N1 influenza virus infection, we challenged IL-17 KO mice on a C57BL/6 background and C57BL/6 wild-type controls with 10LD50 VN1194. Following viral challenge, IL-17 KO mice showed a marked reduction in body weight during the course of H5N1 infection (Figure 1b), which was closely correlated with the significantly lower survival rate observed in IL-17 KO mice (Figure 1a). These data suggest that IL-17 may play a protective role in the immune response against lethal H5N1 influenza virus infection. To determine whether decreased viral clearance correlates with increased mortality in IL-17 KO mice, we measured the viral burden in the lung tissue of mice at 5 days post-infection (dpi) by real-time PCR using primers for the viral NP gene. Compared with wild-type controls, IL-17 KO mice exhibited a significantly higher viral burden in the lung at 5 dpi (Figure 1c). Taken together, these results indicate that IL-17 plays a protective role during H5N1 influenza virus infection.

IL-17 KO mice infected with H5N1 influenza virus displayed reduced weight and higher mortality. (a, b) Following challenge with 10LD50 influenza virus VN1194, IL-17 KO mice (black lines) showed a significantly lower survival rate (a, n=33 in wild-type group, n=27 in IL-17 KO group) and more weight loss (b, n=4 in each group) compared to wild-type mice (grey lines). (c) Viral load in the lungs at 5 dpi was measured by real-time PCR using primers for the NP gene (n=4 in each group). *P<0.05, ***P<0.001. dpi, days post-infection; KO, knockout; LD50, 50% lethal dose; NP, nucleoprotein.
H5N1 infection causes a significant increase in the severity of pulmonary damage in IL-17 KO mice
To evaluate the pulmonary immunopathology caused by H5N1 infection, semiquantitative histological assessment of inflammation and tissue damage in lungs was performed. This analysis rated immunopathology according to the degeneration and necrosis levels of bronchi and bronchiolar epithelium, the infiltration of inflammatory cells, and alveoli degeneration and collapse. Consistent with their higher morbidity and mortality, H5N1-infected IL-17 KO mice exhibited lung damage with significantly increased severity compared to wild-type controls (Figure 2a and b). Notably, both IL-17 KO mice and wild-type controls showed severe pathology in the lower and upper respiratory tracts, consistent with previous findings demonstrating that the H5N1 virus can attach to both upper and lower respiratory tracts and cause severe pulmonary lesions in mice.29, 30

Histopathological evaluation of lung tissue from IL-17 KO and wild-type mice at 5 dpi following challenge with 10LD50 influenza virus VN1194. (a) Lungs of IL-17 KO mice displayed more severe damage than those of wild-type mice. Lung tissue was collected at 5 dpi and sections were stained with hematoxylin and eosin. Sections are representative of four mice in each group. Images are at magnification ×200. (b) Pathological changes were scored as described in the section on ‘Lung fixation and histopathological assessment’. *P<0.05, **P<0.01. dpi, days post-infection; IL, interleukin; KO, knockout; LD50, 50% lethal dose.
Decreased number of B cells in the lungs of IL-17 KO mice during H5N1 virus infection
During influenza virus infection in humans and other mammals, B cells are recruited from the circulation to the site of infection, i.e., the respiratory tract.31, 32 To determine whether IL-17 affects B-cell responses, we examined the frequencies of B cells in the lung tissue of mice infected with H5N1 virus or stimulated with inactivated H5N1 virus. Immunohistochemical examination of lung tissue from live virus-infected IL-17 KO mice revealed significantly reduced B-cell numbers (Figure 3a). To further evaluate the infiltration of B cells into the lung after infection, we vaccinated the mice with inactivated H5N1 virus and enumerated the total number of immune cells in whole lung tissue (Figure 3b–d). Consistent with the immunohistological findings, a significant decrease in the total number of B cells (i.e. predominantly B220+CD5− B2 cells) in the whole lung of IL-17 KO mice was observed when compared to wild-type control animals (data not shown). Interestingly, reduced numbers and frequencies of dendritic cells (DCs) in the lungs of IL-17 KO mice were also observed with no obvious changes for T cells, natural killer (NK) cells, natural killer T cells or macrophages (MΦ) (Figure 3c and d).

The total number and frequency of B cells were significantly reduced in the lung tissue of IL-17 KO mice upon virus stimulation. (a) Immunohistochemical staining of B220+ B cells in the lungs collected at 5 dpi. Sections are representative of four mice in each group. Images are at magnification ×400. (b) Flow cytometric analysis of B-cell frequencies among immune cells in the lung 48 h post-stimulation with inactivated virus. Numbers indicate the percentage of B220+ cells. Figures are representative of three mice in each group. (c, d) Cellularity analysis of B cells (B220+), T cells (CD3+), γσT cells (γσTCR+), NK cells (NK1.1+CD3-), NKT cells (NK1.1+CD3+), DCs (CD11c+), neutrophils (Gr1+CD11b+) and macrophages (Gr1−CD11b+) in the lungs 48 h post-stimulation with inactivated virus (n=3 in each group). *P<0.05.DC, dendritic cell; dpi, days post-infection; IL, interleukin; KO, knockout; NK, natural killer; NKT, natural killer T.
B cells from IL-17 KO mice display impaired migration capacity
The reduced frequency of B cells in the lungs of virus-challenged IL-17 KO mice prompted us to investigate whether B cells from IL-17 KO mice display any defect in their chemotactic migration to B-lymphocyte chemoattractant (BLC/CXCL13), a homeostatic chemokine highly expressed in the pleural cavity and critically involved in B-cell recruitment during infection.33, 34, 35 In the presence of CXCL13, B cells from wild-type mice showed a higher chemotaxis rate than B cells from IL-17 KO mice in vitro (Figure 4a). CXCL13 (C-X-C motif chemokine 13) is the exclusive ligand for CXCR5 (chemokine receptor type 5).36 Therefore, we examined CXCR5 expression in B cells from IL-17 KO mice after inactivated virus stimulation in vitro. Indeed, B cells from IL-17 KO mice showed reduced CXCR5 expression 2 h after virus stimulation when compared with wild-type controls (Figure 4b and c). Moreover, in vivo experiments demonstrated that B cells from the lungs of IL-17 KO mice exhibited decreased expression of CXCR5 following stimulation with inactivated virus (Figure 4d). Remarkably, we detected an approximate fivefold reduction of CXCL13 mRNA levels in the lung tissue of IL-17 KO mice (Figure 4e). Furthermore, immunohistochemical analysis confirmed more robust staining of CXCL13 protein in the lung tissue of wild-type mice when compared to that of IL-17 KO mice (Figure 4f). Thus, these results suggest that reduced CXCR5 expression in B cells from IL-17 KO mice, together with decreased CXCL13 in the inflammation milieu, leads to a markedly impaired migration capacity of B cells into the lungs of IL-17 KO mice during influenza virus infection.

Reduced B-cell migration to the lung in IL-17 KO mice. (a) Analysis of chemotactic migration of B cells towards CXCL13. Splenic B cells from both IL-17 KO mice and wild-type controls were purified with B220 microbeads and then subjected to a CXCL13 chemotaxis assay. Migrated cells were counted on a flow cytometer under a constant medium flow rate. Each treatment was performed with triplicate transwells. (b, c) CXCR5 expression in B cells from wild-type (gray lines) and IL-17 KO mice (black lines) upon stimulation of inactivated virus in vitro. Isolated splenic B cells were set up at 5×106 cells/ml in R10 with 10 µg/ml inactivated influenza virus VN1194. CXCR5 expression in B cells was evaluated before (0 h), 2 and 7 h after stimulation. Data were obtained in triplicate and expressed as MFI±s.d. detected by flow cytometry. (d) CXCR5 expression in B cells from the lungs of mice stimulated with inactivated virus in vivo. Both IL-17 KO mice and wild-type controls were stimulated i.n. with inactivated virus. Mice were killed at 48 h post-stimulation, and surface expression of CXCR5 in B cells from the lungs was analyzed by flow cytometry. Data were obtained from three animals in each group and expressed as MFI±s.d., as determined by flow cytometric analysis. (e, f) CXCL13 expression in the lung tissue of IL-17 KO mice and wild-type mice following H5N1 influenza virus infection. Mice were infected with H5N1 influenza virus and mRNA or protein levels of CXCL13 in lung tissue collected 5 dpi were evaluated by real-time PCR (e) and immunohistochemical staining (f), respectively (n=4 in each group). *P<0.01, **P<0.05. dpi, days post-infection; IL, interleukin; i.n., intranasally; KO, knockout; MFI, mean fluorescence intensity.
Discussion
In this study, we have shown that H5N1-infected IL-17 KO mice display substantially reduced weight, more severe lung immunopathology and significantly reduced survival compared to wild-type controls. Moreover, we have detected dramatically decreased B-cell numbers in the lungs of IL-17 KO mice after virus infection, which might contribute to severely compromised pulmonary virus clearance. Our findings have demonstrated that IL-17 plays a protective role during high pathogenic H5N1 infection via modulating the B-cell response, an important arm of adaptive immunity.
A growing body of evidence indicates that IL-17, a key pro-inflammatory cytokine, positively associates with the development of several autoimmune diseases.7 Although pro-inflammatory factors are critical mediators of autoimmune responses, they can act as protective factors in eliciting an effective immune response during infection. It has been established that IL-17 mediates protection during the immune response against extracellular bacterial and fungal infection in the lung.37, 38 The most important feature of the highly pathogenic H5N1 influenza virus infection is the exaggerated cytokine production, also called a ‘cytokine storm’, which has been evaluated in many studies. However, while IL-17 was reported to be relevant to poor outcomes in seasonal and pandemic H1N1,16, 17 little is known if IL-17 has a detrimental or beneficial role in the highly pathogenic H5N1 influenza virus infection. We have clearly demonstrated that IL-17 plays a beneficial role in the immune response to H5N1 influenza infection and in viral clearance.
To further elucidate the underlying mechanisms of the observed protective role of IL-17, we assessed the immune response in the lungs of wild-type and IL-17 KO mice. As influenza virus infection is predominantly pulmonary-restricted, we performed an analysis primarily focused on the response in this inflammatory site. The decreased frequency and number of B cells in the lungs of IL-17 KO mice indicated a severely impaired B-cell response in these H5N1-infected mice (Figure 3). B cells exert important protective functions throughout the course of infection by participating in antigen presentation and antibody secretion. Recent studies have indicated an important role of B cells in virus clearance that is initiated in the very early phases of influenza virus infection through the production of pre-existing natural antibodies and neutralizing antibodies as early as 2 dpi.32, 39 Efficient virus clearance is dependent on both the cellular and humoral arms of the immune system being intact. The decrease in B-cell numbers at the site of inflammation in infected IL-17 KO mice is consistent with their increased viral load and the increased severity of immunopathology. Thus, it is reasonable to speculate that an impaired B-cell response may partially account for the increased morbidity and mortality of the IL-17 KO mice upon infection with H5N1 (Figure 1a and b). Interestingly, the phenotypes observed in this study are similar to the pathological changes described for fatal human H5N1 influenza infection.40 These data implicate a deficiency in antibody production in IL-17 KO mice,24 which may be an important factor in their weakened immunity to H5N1 influenza infection. Thus, future studies are needed to determine the contribution of both IgM and neutralizing antibody production to viral clearance in the lungs of IL-17 KO mice during H5N1 infection. Although the cellular source of IL-17 during H5N1 infection remains to be identified, recent studies suggest that γσT cells and CD8+ T cells may contribute to early IL-17 production during influenza infection.12, 16 However, the in vivo role of IL-17 in the clearance of influenza virus and its immunopathology has been controversial. Several studies, including ours, have demonstrated the important protective role of IL-17 against lethal flu infection.12, 13, 41 In contrast, other groups have showed that IL-17 signaling was not required for viral clearance, but rather, played a pathogenic role by recruiting neutrophils to the site of inflammation.16, 42, 43 These apparent discrepancies may be reconciled by strain differences between the various mouse models and subtypes/strains of influenza virus used in this work versus previous studies. In addition to the well-recognized notion that immune responses against H5N1 infection are different from those observed during H1N1 infection, a recent study using IL-10 KO mice has demonstrated that cytokines may exert different effects during sublethal versus lethal influenza infection.13
In vivo and in vitro studies have demonstrated that IL-17 alone drives germinal center formation and antibody class switching.24, 44 IL-17 is also reported to participate in chronic gastric inflammation by modulating B-cell recruitment23, 33, 44 and in autoimmune disease by promoting the production of pathogenic autoantibodies.45 Those reports indicate a potential role for IL-17 in modulating B cells during infection. Consistent with this notion, we first detected impaired B-cell recruitment in the lungs of IL-17 KO mice with H5N1 influenza virus infection. Tissue-specific homing of B cells is modulated by chemokine production at the site of inflammation. CXCR5 and its ligand CXCL13 are essential for B-cell migration within a specific anatomical compartment, thus promoting local immunity to influenza.33, 44, 46, 47 Previous studies have demonstrated that IL-17 directly induced CXCL13 production in the lung tissue and this function was impaired in IL-17A KO mice.48 In line with these findings, we also detected significantly reduced levels of CXCL13 transcripts in the lung tissue of influenza virus-infected IL-17 KO mice compared to wild-type controls (Figure 4e and f). Recent studies have indicated that several cell types contribute to CXCL13 production in the lung tissue during influenza virus infection, including resident macrophage,49 DCs,50 Th17 and pulmonary fibroblast cells.48 Therefore, in addition to the complete absence of IL-17, the reduced frequencies and numbers of DCs in the lung tissue of IL-17 KO mice, as observed in this study, might contribute to reduced CXCL13 production. IL-17, in synergism with granulocyte–macrophage colony-stimulating factor, has been shown to greatly promote both DC proliferation and function;51 thus, it remains to be determined whether IL-17 deficiency affects DC proliferative expansion or contributes to the reduced number of DCs in the lung upon H5N1 infection. Because CXCL13 is a potent B-cell chemoattractant, it is highly likely that decreased CXCL13 expression leads to the observed decrease in B-cell frequency in IL-17 KO mice. Furthermore, both in vitro (Figure 4a–c) and in vivo (Figure 4d) experiments showed impaired B-cell chemotaxis towards CXCL13 and decreased surface expression of CXCR5, which indicates that B cells in wild-type and IL-17 KO mice might differ in their responsiveness to the chemokine CXCL13. Nevertheless, further studies are warranted to identify the cellular source of CXCL13 during H5N1 infection.
During severe H5N1 infection in humans, the uncontrolled virus-induced cytokine storm is believed to be the primary cause of death. Our findings have provided new evidence of a role for IL-17 in modulating the recruitment of B cells during viral infection. Further studies may facilitate the development of new therapeutic strategies for the treatment of H5N1 influenza virus infection in humans.
References
Yuen KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet 1998; 351: 467–471.
Oxford JS . Influenza A pandemics of the 20th century with special reference to 1918: virology, pathology and epidemiology. Rev Med Virol 2000; 10: 119–133.
Gambotto A, Barratt-Boyes SM, de Jong MD, Neumann G, Kawaoka Y . Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008; 371: 1464–1475.
Peiris JS, Poon LL, Guan Y . Emergence of a novel swine-origin influenza A virus (S-OIV) H1N1 virus in humans. J Clin Virol 2009; 45: 169–173.
Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA 2008; 105: 8091–8096.
Osterholm MT . Proposed mechanism of the cytokine storm evoked by influenza virus. N Engl J Med 2005; 352: 1839.
Lai Kwan Lam Q, King Hung Ko O, Zheng BJ, Lu L . Local BAFF gene silencing suppresses Th17-cell generation and ameliorates autoimmune arthritis. Proc Natl Acad Sci USA 2008; 105: 14993–14998.
O'Quinn DB, Palmer MT, Lee YK, Weaver CT . Emergence of the Th17 pathway and its role in host defense. Adv Immunol 2008; 99: 115–163.
Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med 2005; 202: 761–769.
Kohyama S, Ohno S, Isoda A, Moriya O, Belladonna ML, Hayashi H et al. IL-23 enhances host defense against vaccinia virus infection via a mechanism partly involving IL-17. J Immunol 2007; 179: 3917–3925.
Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette–Guerin infection. J Immunol 2007; 178: 3786–3796.
Hamada H, Garcia-Hernandez Mde L, Reome JB, Misra SK, Strutt TM, McKinstry KK et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol 2009; 182: 3469–3481.
McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol 2009; 182: 7353–7363.
Norton EB, Clements JD, Voss TG, Cardenas-Freytag L . Prophylactic administration of bacterially derived immunomodulators improves the outcome of influenza virus infection in a murine model. J Virol 2010; 84: 2983–2995.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6: 1133–1141.
Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol 2009; 183: 5301–5310.
Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramirez P et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care 2009; 13: R201.
Kohlmeier JE, Woodland DL . Immunity to respiratory viruses. Annu Rev Immunol 2009; 27: 61–82.
Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J . B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 2000; 192: 271–280.
Harada Y, Muramatsu M, Shibata T, Honjo T, Kuroda K . Unmutated immunoglobulin M can protect mice from death by influenza virus infection. J Exp Med 2003; 197: 1779–1785.
Palladino G, Mozdzanowska K, Washko G, Gerhard W . Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice. J Virol 1995; 69: 2075–2081.
Xie S, Li J, Wang JH, Wu Q, Yang P, Hsu HC et al. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol 2010; 184: 2289–2296.
Algood HM, Allen SS, Washington MK, Peek RM Jr, Miller GG, Cover TL . Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J Immunol 2009; 183: 5837–5846.
Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA 2010; 107: 14292–14297.
Reed LJ, Muench H . A simple method of estimation fifty percent endpoints. Am J Hyg 1938; 27: 493–497.
Sun S, Zhao G, Xiao W, Hu J, Guo Y, Yu H et al. Age-related sensitivity and pathological differences in infections by 2009 pandemic influenza A (H1N1) virus. Virol J 2011; 8: 52.
van den Brand JM, Stittelaar KJ, van Amerongen G, Rimmelzwaan GF, Simon J, de Wit E et al. Severity of pneumonia due to new H1N1 influenza virus in ferrets is intermediate between that due to seasonal H1N1 virus and highly pathogenic avian influenza H5N1 virus. J Infect Dis 2010; 201: 993–999.
Sauer KA, Scholtes P, Karwot R, Finotto S . Isolation of CD4+ T cells from murine lungs: a method to analyze ongoing immune responses in the lung. Nat Protoc 2006; 1: 2870–2875.
van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD et al. H5N1 virus attachment to lower respiratory tract. Science 2006; 312: 399
Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med 2007; 13: 147–149.
Coro ES, Chang WL, Baumgarth N . Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J Immunol 2006; 176: 4343–4351.
Rothaeusler K, Baumgarth N . B-cell fate decisions following influenza virus infection. Eur J Immunol 2010; 40: 366–377.
Ansel KM, Harris RB, Cyster JG . CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 2002; 16: 67–76.
Hopken UE, Achtman AH, Kruger K, Lipp M . Distinct and overlapping roles of CXCR5 and CCR7 in B-1 cell homing and early immunity against bacterial pathogens. J Leukoc Biol 2004; 76: 709–718.
Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J . Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 2006; 107: 2613–2618.
Ebert LM, Schaerli P, Moser B . Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol 2005; 42: 799–809.
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 2001; 194: 519–527.
Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol 2003; 170: 4432–4436.
Baumgarth N, Choi YS, Rothaeusler K, Yang Y, Herzenberg LA . B cell lineage contributions to antiviral host responses. Curr Top Microbiol Immunol 2008; 319: 41–61.
Taubenberger JK, Morens DM . The pathology of influenza virus infections. Annu Rev Pathol 2008; 3: 499–522.
Norton EB, Clements JD, Voss TG, Cardenas-Freytag L . Prophylactic administration of bacterially derived immunomodulators improves the outcome of influenza virus infection in a murine model. J Virol 2010; 84: 2983–2995.
Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010; 115: 335–343.
Linden A, Laan M, Anderson GP . Neutrophils, interleukin-17A and lung disease. Eur Respir J 2005; 25: 159–172.
Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD . Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci USA 2007; 104: 10577–10582.
Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 2008; 9: 166–175.
Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M . A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 1996; 87: 1037–1047.
Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT . A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature 1998; 391: 799–803.
Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol 2011; 12: 639–646.
Hojgaard A, Close R, Dunn DM, Weiss RB, Weis JJ, Weis JH . Altered localization of CXCL13 expressing cells in mice deficient in Pactolus following an inflammatory stimulus. Immunology 2006; 119: 212–223.
GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med 2009; 206: 2339–2349.
Liu B, Tan W, Barsoum A, Gu X, Chen K, Huang W et al. IL-17 is a potent synergistic factor with GM-CSF in mice in stimulating myelopoiesis, dendritic cell expansion, proliferation, and functional enhancement. Exp Hematol . 2010; 38: 877 884. e1.
Acknowledgements
We are grateful to Dr Yoichiro Iwakura (The University of Tokyo) for providing IL-17 KO mice. This work was supported by the Research Fund for the Control of Infectious Diseases (RFCID), Food and Health Bureau, Hong Kong SAR Government (No. 10091002). The authors declare no financial or commercial conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, X., Chan, C., Yang, M. et al. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell Mol Immunol 8, 462–468 (2011). https://doi.org/10.1038/cmi.2011.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2011.38
Keywords
This article is cited by
-
Baseline innate and T cell populations are correlates of protection against symptomatic influenza virus infection independent of serology
Nature Immunology (2023)
-
Role of tertiary lymphoid organs in the regulation of immune responses in the periphery
Cellular and Molecular Life Sciences (2022)
-
The HSP70-fused foot-and-mouth disease epitope elicits cellular and humoral immunity and drives broad-spectrum protective efficacy
npj Vaccines (2021)
-
Host-derived lipids orchestrate pulmonary γδ T cell response to provide early protection against influenza virus infection
Nature Communications (2021)
-
Oral immunization with a novel attenuated Salmonella Typhimurium encoding influenza HA, M2e and NA antigens protects chickens against H7N9 infection
Veterinary Research (2018)
