
Abstract
Historically, influenza pandemics have arisen from avian influenza viruses. Avian influenza viruses H5N1 and H9N2 are potential pandemic candidates. Infection of humans with the highly pathogenic avian influenza H5N1 virus is associated with a mortality in excess of 60%, which has been attributed to dysregulation of the cytokine system. Human macrophages and epithelial cells infected with some genotypes of H5N1 and H9N2 viruses express markedly elevated cytokine and chemokine levels when compared with seasonal influenza A subtype H1N1 virus. The mechanisms underlying this cytokine and chemokine hyperinduction are not fully elucidated. In the present study, we demonstrate that autophagy, a tightly regulated homeostatic process for self-digestion of unwanted cellular subcomponents, plays a role in cytokine induction. Autophagy is induced to a greater extent by H9N2/G1, in association with cytokine hyperinduction, compared with H1N1 and the novel pandemic swine-origin influenza A/H1N1 viruses. Using 3-methyladenine to inhibit autophagy and small interfering RNA to silence the autophagy gene, Atg5, we further show that autophagic responses play a role in influenza virus-induced CXCL10 and interferon-α expression in primary human blood macrophages. Our results provide new insights into the pathogenic mechanisms of avian influenza viruses.
Similar content being viewed by others


Introduction
Avian influenza viruses pose both pandemic and zoonotic threats to humans. Human H5N1 disease is associated with mortality in excess of 60%,1 and is a cause for global public health concerns. In addition, avian influenza H9N2 viruses have become highly prevalent in poultry in many countries and have caused repeated cases of human infections.2, 3 Studies exploring the mechanism of transmission have shown that many of these H9N2 isolates have the capacity to bind to both sialic acid (SA) α 2-3 and SA α 2-6 receptors found on avian and human cells, respectively. These leucine-226-containing H9N2 viruses with human cell tropism grew efficiently in human airway epithelial cells4 and were efficiently transmitted in ferrets.5 Additionally, replication in ferrets and transmission via direct contact were significantly enhanced in an H9N2 avian-human reassortant virus, although no aerosol transmission was detected.5 Taken together, the high prevalence, wide geographic range, rapid evolution and frequent reassortment of H9N2 viruses in poultry,6 in combination with the increasing number of avian influenza infections in humans, highlight their pandemic and zoonotic potential.
Due to their potential threat as pandemic pathogens, details of avian virus replication, pathogenesis and host response are subjects of intense investigation. Previous reports, including ours,7, 8 demonstrated differences in the pattern of cell death in avian H5N1 and human influenza virus-infected cells. In response to insults, cell death processes include necrosis, apoptosis and autophagy. Apoptotic death is an organized process to minimize inflammatory changes, whereas autophagy is a self-controlled program for eliminating damaged organelles. Autophagy was originally characterized as a tightly regulated cellular homeostatic process, which involves the sequestration of parts of the cytosol and intracellular organelles within double-membraned autophagic vacuoles and their subsequent delivery to lysosomes for degradation. In addition to cell survival, autophagy plays an essential role in innate and adaptive immunity to fight against microbial infections.9 For example, autophagy enhances degradation of HSV-1 viral proteins,10 as well as recognition of single-stranded RNA viruses and secretion of interferon-α (IFN) in plasmacytoid dendritic cells.11 Autophagy is also important in lysosomal proteolysis of foreign antigens and their presentation by major histocompatibility complex class II molecules to CD4+ cells. Autophagosomes have been shown to fuse continuously with multivesicular major histocompatibility complex class II-loading compartments.12 The role of autophagy in cellular antiviral responses is not well defined. However, various viruses, including poliovirus, rotavirus and parvovirus, can induce autophagy to enhance their own replication.13
Fatal outcomes of H5N1 virus infections are associated with high viral load and hypercytokinemia.14 Elevated levels of CXCL10, CXCL9 and CCL2 were found in patients with H5N1 infections when compared to those with human seasonal influenza H3N2 and H1N1 virus infections. In other in vitro studies of primary human macrophages, the highly pathogenic H5N1 (A/Hong Kong/483/1997) and H9N2/G1 (A/Quail/Hong Kong/G1/97) viruses induced significantly higher levels of cytokines and chemokines, including tumor necrosis factor-α, type I IFN, CCL2, CCL3, CCL5 and CXCL10, when compared with the H1N1 virus.15, 16 These two avian viruses share common internal viral genes.17 A recent report showed that the novel swine-origin influenza A/H1N1 virus (S-OIV), which caused the recent pandemic, induced cytokine and chemokine levels comparable with the seasonal H1N1 virus in primary human blood macrophages.18 Because autophagy plays a role in influenza virus-induced cell death,19 it may also contribute to cytokine dysregulation. Here, we show that while influenza viruses H1N1 (both seasonal and pandemic) and H9N2/G1 induce autophagy in human blood macrophages, H9N2 viruses are far more potent inducers of autophagy. In addition, we demonstrate that S-OIV is a weaker inducer of autophagy than the seasonal H1N1 virus. We also examine the role of autophagy in CXCL10 and IFN-α production upon influenza virus infection.
Materials and methods
Reagents and antibodies
Specific antibodies against LC3B, Atg5 and phosphorylated and unphosphorylated forms of p70S6 kinase (p70S6K) were purchased from Cell Signaling Technology (Beverly, MA, USA), while antibodies specific to p62 and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). We purchased 3-methyladenine (3-MA) from Sigma (St Louis, MO, USA). Small interfering RNA (siRNA) oligos specific to Atg5 (siAtg5) and non-targeting siRNA oligos (siCtrl) were purchased from Dharmacon (Lafayette, CO, USA).
Cells and viruses
Human blood macrophages from healthy donors (Hong Kong Red Cross Blood Transfusion Service) were prepared as previously described.7, 20, 21 In brief, blood mononuclear cells were separated by Ficoll–Paque centrifugation and purified by the adherence method. Monocytes were seeded in 24-well (0.5×106 cells/well) or 6-well (2×106 cells/well) tissue culture plates for studies of RNA and protein, respectively. Monocytes were differentiated in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 5% heat-inactivated autologous plasma. Differentiated macrophages were obtained after culturing for 14 days. Influenza viruses including A/Quail/Hong Kong/G1/97 (H9N2/G1), A/Hong Kong/54/98 (H1N1) and the pandemic swine-origin influenza virus A/HongKong/415742/09 (S-OIV) were grown in Madin–Darby canine kidney cells and were purified by pre-adsorption to and elution from turkey red blood cells. Virus infectivity was determined by titration on Madin–Darby canine kidney cells.7, 20
Virus infections
Macrophages were infected with the viruses at a multiplicity of infection of 2 for 30 min at 37 °C. The supernatant containing the virus inoculum was then removed, and the cells were incubated in macrophage serum-free medium (Invitrogen) supplemented with 0.6 µg/ml penicillin and 60 µg/ml streptomycin. The mock-treated control was incubated with the buffer under parallel conditions.
Protein extraction and western blot analysis
Whole-cell lysates were prepared with total lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 50 mM NaF, 10 mM β-glycerophosphate, 0.1 mM EDTA, 10% glycerol, 1% Triton X-100) containing a protease inhibitor cocktail. For western blot analysis, 30 µg of protein was heat denatured in sample buffer (125 mM Tris, pH 6.8, 4% sodium dodecyl sulfate, 20% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue), separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto a nitrocellulose membrane for assaying protein levels with ECLTM Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ, USA) solution.
Immunocytochemical staining
For the detection of autophagosome formation, cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X-100 for 5 min. The cells were stained with antibodies specific to LC3B and rhodamine-conjugated antirabbit secondary antibodies. Nuclei were counterstained with 4,6-diamidine-2-phenylindole dihydrochloride, and the immunocytochemical staining was examined under fluorescence microscopes.
siRNA transfections
Expression of Atg5 in human blood macrophages was knocked down by siAtg5. siCtrl were used as controls. Macrophages were transfected with either the siAtg5 or siCtrl at the indicated concentrations using jetPEI transfection reagent (PolyPlus Transfection, Illkirch, France). At 48 h after transfection, cells were infected with the viruses, and cells were harvested at indicated time points for further analyses.
Quantitative reverse transcription-PCR analyses
Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The cDNA was synthesized from total RNA with oligo(dT) primers and Superscript II reverse transcriptase (Invitrogen). The levels of mRNA encoding CXCL10 and IFN-α1, -α2 and -α8 were assayed with TaqMan gene expression assays (Applied Biosystems, Foster City, CA, USA).
Quantitative analyses of CXCL10 and IFN-α by enzyme-linked immunosorbent assays (ELISA)
Supernatant samples of macrophage cultures were collected at the indicated time points after infection and irradiated with UV light before the levels of CXCL10 and IFN-α were measured with specific ELISA Kits (R&D Systems, Minneapolis, MN, USA).
Results
Differential induction of autophagy by H9N2/G1, H1N1 and S-OIV influenza viruses
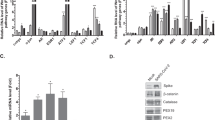
Conversion of LC3B I to II, a hallmark of autophagy,22 was analyzed by western blot. Primary blood macrophages were infected with influenza viruses, including H9N2/G1, H1N1 and S-OIV, at a multiplicity of infection of 2. At 19 h post-infection (h.p.i.), whole-cell lysates were prepared and analyzed by western blot using antibodies specific to LC3B. Actin was used as a loading control. LC3B I was converted to LC3B II at a higher efficiency in cells infected with H9N2/G1 virus than with H1N1 virus (Figure 1a). The intensity ratio of LC3B II to I was determined by measuring the band intensity using Bio-Rad Quantity One imaging software. When compared to mock-treated cells, the ratio of LC3B II to I was increased fivefold upon H1N1 infection and, much more drastically, 90-fold with H9N2/G1. However, when comparing the LC3B II to I ratio in H1N1 versus S-OIV virus-infected cells at 17 h.p.i., the ratio was smaller in the case of S-OIV (Figure 1b), indicating that S-OIV induced less autophagy than H1N1. Autophagosomes were also examined by immunocytochemistry at 19 h.p.i. Consistent with the western blots, LC3B-positive vesicles with a punctate-staining pattern were found in cells infected with H9N2/G1 or H1N1 (Figure 1c). Moreover, more prominent staining was found in cells infected with H9N2/G1 when compared with those infected with H1N1.

Autophagy was induced by H1N1, H9N2/G1 and S-OIV influenza viruses. Human blood macrophages were mock-infected or infected with H1N1, H9N2/G1 or S-OIV. (a, b) Whole-cell lysates were harvested and LC3B expression was examined by western blot analysis. Actin was used as a loading control. (c) Autophagosomes were examined by staining the mock-, H1N1- or H9N2/G1-infected cells with antibodies specific to LC3B. DAPI was used to stain nuclei. Arrows indicate LC3B positive vesicles. Magnification: ×400. (d) p62 expression levels were examined at 19 h.p.i. by western blot analysis. (e) At 2 h.p.i., phosphorylation levels of p70S6K (Thr389) were examined by western blot analysis. Total p70S6K was used as a loading control. Densities of the protein bands were determined with Bio-Rad Quantity One imaging software. The values in parentheses represent LC3B II to LC3B I ratios (a, b). The p62 (d) and phospho-p70S6K (Thr389) (e) intensity values were normalized to the corresponding actin or total p70S6K values. The values in parentheses are the relative normalized intensities compared to those of the mock-treated cells. The results shown are representative figures from three independent experiments. DAPI, 4,6-diamidine-2-phenylindole dihydrochloride; h.p.i., hour post-infection; p70S6K, p70S6 kinase; S-OIV, swine-origin influenza A/H1N1 virus; Thr389, threonine 389.
Additionally, we examined other markers of autophagy, including the level of p62 protein, a polyubiquitin-binding protein and the activity of p70S6K, a marker of mammalian target of rapamycin (mTOR), following influenza virus infections.23, 24 The p62 protein can bind directly to LC3 proteins that enhance the degradation of ubiquitinated proteins in the lysosome through the autophagic processes.24 Because the p62 protein itself can also be degraded by autophagy, its expression level indicates the autophagic status of the cells.25, 26 Our results show that p62 expression decreased by approximately 45% in macrophages with H1N1 infection when compared with mock-treated macrophages (Figure 1d). Strikingly, p62 protein expression was decreased by about 84% in H9N2/G1-infected cells.
Autophagy is negatively regulated by mTOR.27 Inhibition of mTOR leads to induction of autophagy. Because p70S6K is a direct target of mTOR, we examined the phosphorylation of p70S6K at threonine 389 to determine the activity of the rapamycin-sensitive mTOR complex 1.28 Our results show that phosphorylation of p70S6K at threonine 389 in H9N2/G1-infected macrophages decreased at 2 h.p.i. when compared with mock-treated cells (Figure 1e). In contrast, there was no significant change in H1N1-infected cells. Taken together, these results demonstrate that the H9N2/G1 virus induced more autophagic responses than H1N1.
Reduced H9N2/G1- and H1N1-induced autophagy after knockdown of Atg5
To delineate the mechanisms of autophagic induction following H1N1 and H9N2/G1 infections, the autophagic pathways were blocked by knocking down Atg5 expression through siRNA-mediated gene silencing. Atg5 is involved in one of the two ubiquitin-like conjugation systems mediating vesicle elongation during the autophagosome formation process.29 Here, primary macrophages were transfected with siAtg5 or siCtrl at concentrations of 100 or 200 nM. Knockdown efficiency was examined by western blot with antibodies specific to Atg5 at 48 h.p.i. As shown in Figure 2, Atg5 expression was decreased in a dose-dependent manner following administration of siAtg5. For example, the Atg5 level was decreased by more than 70% with siAtg5 at 200 nM when compared with cells transfected with siCtrl.

Knockdown of Atg5 protein by siRNA oligos. Human blood macrophages were transfected with siAtg5 or siCtrl at 100 and 200 nM. At 48 hour post-transfection, whole-cell lysates were harvested and Atg5 expression was measured by western blot with antibodies specific to Atg5. Actin was used as a loading control. Densities of the protein bands were determined with Bio-Rad Quantity One imaging software. The Atg5 intensity values were normalized to the corresponding actin levels. The values in parentheses are the relative normalized intensities compared to those of the siCtrl-transfected cells. The results shown are representative figures from three independent experiments. siAtg5, small interfering RNA oligos specific to Atg5; siCtrl, non-targeting control oligos; siRNA, small interfering RNA.
Following siRNA treatment, Atg5 knockdown macrophages were infected with H1N1 or H9N2/G1 viruses. In cells transfected with siCtrl, increases of LC3B II levels were consistent with the non-transfected cells with H1N1 or H9N2/G1 infections (comparing Figures 1a and 3). However, in cells with reduced levels of Atg5, neither H1N1 nor H9N2/G1 viruses significantly increased LC3B II levels when compared with the mock-treated cells (Figure 3, lanes 2, 4 and 6). Therefore, our results show that influenza virus-induced autophagy is an Atg5-dependent process.

Involvement of Atg5 in H1N1 and H9N2/G1 virus-induced autophagy. siAtg5- or siCtrl-transfected macrophages were mock-infected or infected with H1N1 or H9N2/G1. Atg5 and LC3B II levels were examined by western blot. Actin was used as a loading control. Densities of the protein bands were determined with Bio-Rad Quantity One imaging software. The values in parentheses are the relative intensities of Atg5 or LC3B II levels compared to those of actin. The results shown are representative figures from three independent experiments. siAtg5, small interfering RNA oligos specific to Atg5; siCtrl, non-targeting control oligos; siRNA, small interfering RNA.
Hyperinductions of CXCL10 and IFN-α by H9N2/G1 virus
In response to influenza virus infection, host cells secrete cytokines, chemokines and IFNs as antiviral mediators. H9N2/G1 virus has been shown to induce a cytokine profile similar to that of the highly pathogenic H5N1 virus, and both of these viruses are known to induce significantly higher cytokine levels than H1N1 viruses.15 We further investigated whether autophagy is involved in the hyperinduction of CXCL10 and IFN-α by H9N2/G1. At 3 h.p.i. with H9N2/G1 or H1N1, mRNA levels of CXCL10 were quantitatively analyzed by TaqMan gene expression assays. H9N2/G1 induced significantly higher levels of CXCL10 mRNA when compared with H1N1 (Figure 4a). Furthermore, the levels of CXCL10 in the culture supernatants of virus-infected cells at 8 h.p.i. were measured by CXCL10-specific ELISA (R&D Systems). Consistent with the mRNA results, H9N2/G1 induced much higher levels of CXCL10 protein in the culture supernatants when compared with H1N1 (Figure 4b). Similar to CXCL10, the levels of IFN-α in the cell culture supernatants were increased with H9N2/G1 infection (Figure 4c).

H9N2/G1 induced higher CXCL10 and IFN-α levels than H1N1. Macrophages were either mock-infected or infected with H1N1 or H9N2/G1. (a) At 3 h.p.i., CXCL10 levels were examined by TaqMan gene expression assay; n=6. (b) CXCL10 levels in culture supernatants harvested 8 h.p.i. were measured by ELISA; n=4. (c) IFN-α levels in culture supernatants harvested 8 h.p.i. were measured by ELISA; n=6. *P<0.05. BD, below detection limit; h.p.i., hour post-infection; IFN, interferon.
Involvement of autophagy in CXCL10 and IFN-α production upon influenza virus infection
Autophagy is required for the transport of cytosolic viral replication intermediates into the lysosome for recognition by Toll-like receptor (TLR) 7, which in turn leads to IFN secretion.11 Therefore, we next investigated whether autophagy plays a role in chemokine induction by influenza virus infection. To block the autohpagic pathway we treated cells with 3-MA, a well-known autophagy inhibitor. Cells were treated with 3-MA at 10 mM for 30 min before virus infections, and the levels of CXCL10 mRNA were examined 3 h.p.i. We found that, in the presence of 3-MA, CXCL10 mRNA expression was decreased by more than 80% by both H1N1 and H9N2/G1 infection (Figure 5a and b).

Inhibition of autophagy suppressed influenza-induced CXCL10 and IFN-α expression. Macrophages were pre-treated with 3-MA (10 mM) before infection with H1N1 (a) or H9N2/G1 (b). CXCL10 mRNA expression at 3 h.p.i. was examined by TaqMan gene expression assay; n=3. Macrophages were transfected with siAtg5 or siCtrl at 200 nM. At 48 hour post-transfection, cells were infected with H1N1 (c) or H9N2/G1 (d). The relative CXCL10 mRNA levels were examined at 3 h.p.i. by TaqMan gene expression assay; n=3. Relative mRNA levels of IFN-α1, -α2 and -α8 in siRNA-transfected cells with H9N2/G1 infections were examined by TaqMan gene expression assay (e); n=3. Data represent the percentages of CXCL10 or IFN-α expression from 3-MA-treated or siAtg5-transfected cells relative to the mock-treated or siCtrl-transfected cells, respectively. *P<0.05, #P<0.01. h.p.i., hour post-infection; IFN, interferon; siAtg5, small interfering RNA oligos specific to Atg5; siCtrl, non-targeting control oligos; siRNA, small interfering RNA; 3-MA, 3-methyladenine.
We next examined induction of CXCL10 in H1N1- or H9N2/G1-infected macrophages after transfection with siAtg5 or siCtrl. In siAtg5-trasnfected cell at 3 h.p.i., CXCL10 mRNA levels were decreased by 72 and 67% in H1N1- or H9N2/G1-infected cells, respectively, when compared with those transfected with siCtrl (Figure 5c and d). The mRNA levels of IFN-α1, -α2 and -α8 were measured in H9N2/G1-infected cells by TaqMan gene expression assay. Similarly, in cells transfected with siAtg5, the IFN-α1, -α2 and -α8 mRNA levels were significantly decreased, by around 70%. In addition to CXCL10 and IFN-α, previous reports showed that H9N2/G1 induces a number of cytokines and chemokines.16 Therefore, we investigated the role of autophagy in the induction of IFN-β, CCL2, CCL3, CCL5 and tumor necrosis factor-α mRNA by quantitative reverse transcription-PCR. We found that inhibition of the autophagic pathway with siAtg5 significantly suppressed the H9N2/G1-induced expression of IFN-β, CCL3 and tumor necrosis factor-α but not CCL2 and CCL5 (data not shown). However, the strongest suppressions were found in CXCL10 and IFN-α levels.
We also measured CXCL10 and IFN-α protein levels in culture supernatants of Atg5 knockdown cells. At 8 h.p.i. with H9N2/G1, the level of CXCL10 in cell culture supernatants was decreased by about 50% (Figure 6a), concomitant with reduced Atg5 expression. Similarly, IFN-α expression was decreased by approximately 50% in cells transfected with siAtg5 (Figure 6b).

Atg5 knockdown suppressed H9N2/G1-induced CXCL10 and IFN-α protein expression. Macrophages were transfected with siAtg5 or siCtrl at 200 nM. At 48 hour post-transfection, cells were infected with H9N2/G1. Quantities of CXCL10 (a) (n=4) and IFN-α (b) (n=5) in the culture supernatants at 8 h.p.i. were measured by ELISA. The data represent the percentage of CXCL10 and IFN-α expression from siAtg5-transfected cells relative to siCtrl-transfected cells. *P<0.05, #P<0.01. h.p.i., hour post-infection; IFN, interferon; siAtg5, small interfering RNA oligos specific to Atg5; siCtrl, non-targeting control oligos.
In summary, our results show that autophagy plays a role in H1N1- and H9N2/G1-induced CXCL10 and IFN-α production in human macrophages. The differential induction of autophagy by H1N1 and H9N2/G1 may contribute, in part, to the differences in the CXCL10 and IFN-α production by these two viruses in human macrophages.
Discussion
Influenza viruses of avian origin continue to pose pandemic threats to human health. The ability of these viruses to cross the species barrier from poultry to humans increases their potential to cause the next pandemic. In addition to the highly pathogenic H5N1 influenza viruses, H9N2 viruses have been circulating in poultry and were sporadically introduced into humans through close contact.2, 3 Although the H9N2 viruses cause mild symptoms when compared with the H5N1 counterparts, their rapid evolution6 and ability to infect humans raise great concerns.
In response to influenza virus infections, epithelial cells and leukocytes produce proinflammatory, chemotactic and immunoregulatory cytokines. Chemokines belong to a family of chemotactic cytokines that direct the movement of circulating leukocytes to sites of inflammation, thereby contributing to disease pathogenesis.30 Previous studies have shown that H5N1 infections induce cytokine and chemokine dysregulation leading to systemic tissue injury. Elevated serum levels of chemokines were found in patients with H5N1 infections,14, 31 especially in those with fatal outcomes.14 In vitro studies also showed that H5N1 virus induced significantly higher levels of chemokines in human macrophages than H1N1 virus.15, 16 The H9N2/G1 virus used in this report, A/Quail/Hong Kong/G1/97, which shares six internal genes with A/Hong Kong/483/1997 (H5N1),17 was found to induce cytokine and chemokine levels in monocyte-derived macrophages similar to H5N1. In contrast, the novel pandemic S-OIV induced cytokine and chemokine levels similar to the seasonal H1N1 virus in primary human blood macrophages.18 Consistent with previous studies, CXCL10 and IFN-α levels were increased upon infection with H9N2/G1 (Figure 4) but not with S-OIV (data not shown). This correlates with our results showing that H9N2/G1 induced a higher level of autophagy when compared to H1N1 and S-OIV.
We further demonstrated that autophagy is involved in induction of CXCL10 and IFN-α with a well-known autophagy inhibitor, 3-MA, and siRNA-mediated gene silencing of Atg5, which is a critical component in autophagosome formation. After inhibiting the autophagic pathway by knocking down Atg5, H9N2/G1-induced CXCL10 and IFN-α protein levels were decreased by 50% (Figure 6). However, the detailed mechanisms underlying how H9N2/G1 induces CXCL10 and IFN-α through autophagy need to be further investigated. One possibility is recognition of viral components through TLR3. TLR3 recognizes double-stranded RNA produced by many viruses during replication and triggers downstream pathways leading to the production of IFN and cytokines.32 For example, TLR3 is important for induction of CXCL10 during respiratory syncytial virus infection,33 while interactions between TLR3 and its ligand can induce autophagy.34
Similarly, autophagy is involved in virus recognition and IFN induction11 via presentation of cytosolic viral replication intermediates into the lysosome for TLR7 recognition. After virus recognition by TLR7, the IFN production cascade is initiated.35 TLR7 was localized in the same intracellular compartments as TLR3.36 H9N2/G1-induced CXCL10 and IFN-α production may be dependent on autophagy for recognition of viral components by TLR3. In addition, CXCL10 induction upon virus infection can be triggered by alternative pathways, including IFN-regulatory factor-3.37 We have previously shown that, when compared to H1N1 virus, the H5N1 viruses are more potent activators of IFN-regulatory factor-3,38 which may explain why knockdown of Atg5 expression in our system could not completely abrogate induction of CXCL10 and IFN-α by H9N2/G1. However, we provide evidence that autophagy contributes, at least in part, to the hyperinduction of CXCL10 and IFN-α during H9N2/G1 infection.
Autophagy and its associated cellular responses are cell type-dependent. In a recent report, it was shown that influenza virus replication is enhanced by autophagy in human lung carcinoma (A549) and canine kidney (Madin–Darby canine kidney) cell lines.19 In the present study, however, blocking autophagy by knocking down Atg5 had no effect on H9N2/G1 virus replication in primary human blood macrophages (data not shown). These results indicate that H9N2/G1 replication is not affected by autophagy in blood macrophages. This discrepancy between human macrophages and lung cancer cells may be due to cell type-specific differences in the autophagic response. Additionally, transformed cell lines may have altered autophagic processes, causing primary cells and transformed cell lines to respond differently to virus infections or other stimuli. A cell type-specific role of autophagy in virus replication has also been demonstrated in coronavirus infections. Autophagy enhances the replication of coronavirus mouse hepatitis virus in embryonic stem cells, and the replication was decreased by more than 99% in Atg5 knockout cells.39 On the other hand, another study demonstrated that Atg5 knockdown had no effect on mouse hepatitis virus replication in primary bone marrow-derived macrophages and primary low-passage mouse embryonic fibroblasts.40 These paradoxical results highlight the importance of tissue origins and cell types in autophagic responses.
To conclude, our present report provides evidence for the involvement of autophagy in influenza virus-induced CXCL10 and IFN-α expression and demonstrates the differential induction of CXCL10 and IFN-α by H9N2 and H1N1. Our results provide new insights into the mechanisms underlying the pathogenesis of avian influenza virus infections.
References
Available from: http://www.who.int/csr/disease/avian_influenza/country/cases_table_2010_03_04/en/index.html
Butt KM, Smith GJ, Chen H, Zhang LJ, Leung YH, Xu KM et al. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J Clin Microbiol 2005; 43: 5760–5767.
Peiris M, Yuen KY, Leung CW, Chan KH, Ip PL, Lai RW et al. Human infection with influenza H9N2. Lancet 1999; 354: 916–917.
Wan H, Perez DR . Amino acid 226 in the hemagglutinin of H9N2 influenza viruses determines cell tropism and replication in human airway epithelial cells. J Virol 2007; 81: 5181–5191.
Wan H, Sorrell EM, Song H, Hossain MJ, Ramirez-Nieto G, Monne I et al. Replication and transmission of H9N2 influenza viruses in ferrets: evaluation of pandemic potential. PLoS One 2008; 3: e2923.
Xu KM, Smith GJ, Bahl J, Duan L, Tai H, Vijaykrishna D et al. The genesis and evolution of H9N2 influenza viruses in poultry from southern China, 2000 to 2005. J Virol 2007; 81: 10389–10401.
Mok CK, Lee DC, Cheung CY, Peiris M, Lau AS . Differential onset of apoptosis in influenza A virus H5N1- and H1N1-infected human blood macrophages. J Gen Virol 2007; 88: 1275–1280.
Zhou J, Law HK, Cheung CY, Ng IH, Peiris JS, Lau YL . Functional tumor necrosis factor-related apoptosis-inducing ligand production by avian influenza virus-infected macrophages. J Infect Dis 2006; 193: 945–953.
Schmid D, Munz C . Innate and adaptive immunity through autophagy. Immunity 2007; 27: 11–21.
Talloczy Z, Virgin HW 4th, Levine B . PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006; 2: 24–29.
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A . Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007; 315: 1398–1401.
Schmid D, Pypaert M, Munz C . Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007; 26: 79–92.
Miller S, Krijnse-Locker J . Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008; 6: 363–374.
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 2006; 12: 1203–1207.
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 2002; 360: 1831–1837.
Zhou J, Law HK, Cheung CY, Ng IH, Peiris JS, Lau YL . Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J Infect Dis 2006; 194: 61–70.
Guan Y, Shortridge KF, Krauss S, Webster RG . Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci USA 1999; 96: 9363–9367.
Woo PC, Tung ET, Chan KH, Lau CC, Lau SK, Yuen KY . Cytokine profiles induced by the novel swine-origin influenza A/H1N1 virus: implications for treatment strategies. J Infect Dis 2010; 201: 346–353.
Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J et al. Autophagy is involved in influenza A virus replication. Autophagy 2009; 5: 321–328.
Lee DC, Cheung CY, Law AH, Mok CK, Peiris M, Lau AS . p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J Virol 2005; 79: 10147–10154.
Yim HC, Li JC, Lau JS, Lau AS . HIV-1 Tat dysregulation of lipopolysaccharide-induced cytokine responses: microbial interactions in HIV infection. Aids 2009; 23: 1473–1484.
Mizushima N, Yoshimori T . How to interpret LC3 immunoblotting. Autophagy 2007; 3: 542–545.
McMahon LP, Choi KM, Lin TA, Abraham RT, Lawrence JC Jr . The rapamycin-binding domain governs substrate selectivity by the mammalian target of rapamycin. Mol Cell Biol 2002; 22: 7428–7438.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC . Autophagy inhibition compromises degradation of ubiquitin–proteasome pathway substrates. Mol Cell 2009; 33: 517–527.
Pursiheimo JP, Rantanen K, Heikkinen PT, Johansen T, Jaakkola PM . Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009; 28: 334–344.
Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL . The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008; 4: 851–865.
Ikenoue T, Hong S, Inoki K . Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol 2009; 452: 165–180.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Charo IF, Ransohoff RM . The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 2006; 354: 610–621.
Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 2004; 363: 617–619.
Kawai T, Akira S . Innate immune recognition of viral infection. Nat Immunol 2006; 7: 131–137.
Rudd BD, Burstein E, Duckett CS, Li X, Lukacs NW . Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J Virol 2005; 79: 3350–3357.
Shi CS, Kehrl JH . MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 2008; 283: 33175–33182.
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C . Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004; 303: 1529–1531.
Nishiya T, Kajita E, Miwa S, Defranco AL . TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J Biol Chem 2005; 280: 37107–37117.
Taima K, Imaizumi T, Yamashita K, Ishikawa A, Fujita T, Yoshida H et al. Expression of IP-10/CXCL10 is upregulated by double-stranded RNA in BEAS-2B bronchial epithelial cells. Respiration 2006; 73: 360–364.
Hui KP, Lee SM, Cheung CY, Ng IH, Poon LL, Guan Y et al. Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. J Immunol 2009; 182: 1088–1098.
Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR . Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004; 279: 10136–10141.
Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007; 3: 581–585.
Acknowledgements
This work was supported by Area of Excellence grants to Malik Peiris, K. Y. Yuen and Allan S. Lau (Grant AoE/M-12/06) from the Research Grants Council of Hong Kong and the Research Fund for Control of Infectious Disease (09080832), as well as grants to Allan S. Lau and Malik Peiris from the Research Grants Council Central Allocation (HKU 1/05C).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Law, AY., Lee, DW., Yuen, KY. et al. Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell Mol Immunol 7, 263–270 (2010). https://doi.org/10.1038/cmi.2010.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2010.25
Keywords
This article is cited by
-
Autophagy is involved in the replication of H9N2 influenza virus via the regulation of oxidative stress in alveolar epithelial cells
Virology Journal (2021)
-
Baicalin mitigated Mycoplasma gallisepticum-induced structural damage and attenuated oxidative stress and apoptosis in chicken thymus through the Nrf2/HO-1 defence pathway
Veterinary Research (2019)
-
Characteristics of human infection with avian influenza viruses and development of new antiviral agents
Acta Pharmacologica Sinica (2013)
-
Autophagy, a new target for disease treatment
Science China Life Sciences (2013)
