
Abstract
Triple-negative breast cancers (TNBCs) have poor clinical outcomes owing to a lack of targeted therapies. Activation of the MEK/MAPK pathway in TNBC has been associated with resistance to conventional chemotherapy and biologic agents and has a significant role in poor clinical outcomes. NV1066, a replication-competent herpes virus, infected, replicated in and killed all TNBC cell lines (MDA-MB-231, HCC1806, HCC38, HCC1937, HCC1143) tested. Greater than 90% cell kill was achieved in more-sensitive lines (MDA-MB-231, HCC1806, HCC38) by day 6 at a multiplicity of infection (MOI) of 0.1. In less-sensitive lines (HCC1937, HCC1143), NV1066 still achieved >70% cell kill by day 7 (MOI 1.0). In vivo, mean volume of flank tumors 14 days after treatment with NV1066 was 57 versus 438 mm3 in controls (P=0.002). NV1066 significantly downregulated p-MAPK activation by 48 h in all cell lines in vitro and in MDA-MB-231 xenografts in vivo. NV1066 demonstrated synergistic effects with a MEK inhibitor, PD98059 in vitro. We demonstrate that oncolytic viral therapy (NV1066) effectively treats TNBC with correlation to decreased MEK/MAPK signaling. These findings merit future studies investigating the potential role of NV1066 as a sensitizing agent for conventional chemotherapeutic and biologic agents by downregulating the MAPK signaling pathway.
Similar content being viewed by others
Introduction
Triple-negative breast cancer (TNBC) is one of the most aggressive forms of breast cancer.1, 2 Although the development of hormonal therapy has changed outcomes of many breast cancers, TNBCs are resistant to hormonal and targeted therapies as they lack estrogen receptor, progesterone receptor and the overexpression of the HER-2 receptor.1, 2 Currently, there is a lack of effective targeted therapy for TNBC and much effort is being placed on investigating novel therapies.
Multiple pathways have been shown to have a role in the intracellular signaling process of triple-negative tumors, including activated epidermal growth factor receptor (EGFR), the mitogen-activated protein kinase (MAPK), mammalian target of rapamycin (mTOR), and the Akt/PI3K (phosphoinositol-3-kinase) pathway.3, 4, 5 The MAPK pathway is a major signal-transduction pathway, consisting of Raf-1, extracellular regulated kinase-1 and 2 and p38 MAPK molecules. These signal molecules are involved in the expression of genes regulating cellular proliferation and apoptosis. For TNBC, MAPK is the most relevant of these pathways. This signaling pathway is activated mostly owing to the overexpression of upstream molecules such as EGFR and mutations involving various genes within the cellular cascade, including Ras, Raf, PI3K or Akt. The Ras/Raf/MAPK pathway has been shown to induce resistance to doxorubicin and paclitaxel through ectopic activation of Raf in breast cancer cells.6, 7 Furthermore, activation of p-MAPK has been associated with resistance to other biologic agents, such as drugs targeting EGFR and mTOR. Therefore, finding therapeutic agents that downregulate the MAPK pathway is crucial in the development of novel targeted therapy for TNBC.
Oncolytic viral therapies have shown great promise in preclinical testing as a novel cancer treatment modality.8 One such promising candidate virus for human therapy is the herpes simplex virus-1 (HSV-1). HSV-1 is a unique and potent oncolytic agent, which has been used in multiple clinical trials and demonstrated favorable safety profiles and promising effects against a wide range of cancers.9, 10, 11, 12, 13, 14, 15 NV1066 is a genetically-engineered form of HSV-1, which has shown promise as an oncolytic agent against >110 cell lines,10 including multiple cancer types, such as mesothelioma,16 pancreatic cancers,17, 18 melanoma,19 and breast cancer.20 Furthermore, NV1066 replicates selectively in cells resistant to apoptosis, and is a derivative of the oncolytic HSV strain NV1020, which has already demonstrated a favorable safety profile in phase I clinical trials.12, 13, 21
NV1066 is specifically designed to exploit cells with increased activation of MAPK signaling. It contains a deletion of the gene γ134.5 that allows for selective replication in cancer cells with high MEK/MAPK phosphorylation.22 We chose this viral construct to test against multiple TNBC cell lines owing to their elevated baseline expression of activated MAPK. In this study, we used NV1066 as a novel cytotoxic agent for treatment of MEK/MPAK-upregulated TNBC, and as a potential adjunct therapy to sensitize TNBC to other targeted agents.
Materials and methods
Cell lines
Human TNBC cell lines MDA-MB-231, HCC1806, HCC38, HCC1937, and HCC1143 were obtained from the ATCC. MDA-MB-231, HCC1806, HCC38, and HCC1143 were maintained in Roswell Park Memorial Institute medium (supplemented with 10 mM Hepes, 2 mM L-glutamine, 1 mM sodium pyruvate, 1.5 g l−1 sodium bicarbonate, 4.5 g l−1 glucose), and HCC1937 cells were cultured in Iscoves medium (supplemented with 2 mM of L-glutamine). Vero cells for viral plaque assay were maintained in Minimum Essential Medium. All media were supplemented with 10% fetal calf serum with penicillin (100 000 U l−1) and streptomycin (100 mg l−1). Cells were cultured in a 5% CO2 humidified incubator at 37 °C.
Virus
NV1066 is a replication-competent, attenuated HSV-1 whose construction has been described in detail previously.23, 24 This virus is derived from the wild-type HSV-1 F-strain backbone, and has single-copy deletions of ICP-4, ICP-0, and γ134.5, which increase tumor selectivity and decrease virulence. NV1066 also contains a transgene for green fluorescent protein (GFP) under the control of a constitutively active cytomegalovirus promoter. Infected cells thus express GFP that can be detected in vitro by fluorescent microscopy. Viral stocks were propagated on Vero cells and titered by standard plaque assay.
GFP expression
Cells were plated at 2 × 104 per well in 96-well plates. After overnight incubation, cells were infected with NV1066 at multiplicity of infection (MOI) of 0.1 and 1.0 plaque-forming units (PFU) ml−1. At regular time intervals (1, 3, and 5 days), cells were examined for GFP expression using an inverted fluorescence microscope (Nikon Eclipse TE300, Nikon, Tokyo, Japan).
Cytotoxicity assays
3 × 104 cells per well of each cell line were plated in 24-well plates overnight. NV1066 was added to each well at varying MOIs of 0.01, 0.1 and 1.0. Media alone was used as negative control. Viral cytotoxicity was expressed as the percentage of surviving cells, which were calculated as the lactate dehydrogenase release of infected samples compared with uninfected control. The intracellular lactate dehydrogenase release following lysis was subsequently measured with a Cytotox 96 kit (Promega, Madison, WI, USA) on a spectrophotometer (EL321e, Bio- Tek Instruments) at 490 nm. All samples were analyzed in triplicate.
Viral plaque assays
After infection of TNBC cells with virus and prior to daily cytotoxicity assay, supernatants of each infected well were collected daily for 5 days and immediately frozen at −80 °C for storage. Serial dilutions of each supernatant samples were made to perform standard viral plaque assays on confluent Vero culture plates.
In vivo xenograft model
Twenty female athymic nude mice (Harlan), ages 6–8 weeks, were subcutaneously injected with 5 × 106 MDA-MB-231 cells into the flank and mice were randomized into two groups for treatment. Xenografts were treated with a single injection of NV1066 (1 × 107 PFU in 100 μl) or PBS (100 μl) 2 weeks after tumor formation. Tumor size (length and width) and mouse weight were recorded three times per week. Tumor volumes were calculated by the equation, V (mm3)=(4/3)*(π)*((a/2)2*(b/2)), a=smallest diameter, b=largest diameter. After 2 weeks of treatment, mice were sacrificed and tumors were harvested and immediately frozen using liquid nitrogen for immunohistochemistry studies.
Immunohistochemistry
Serial sections of 5-μm-thick tumor tissues were cut from cryo-frozen tissues. The immunohistochemistry detection of p-MAPK was performed using the Discovery XT processor (Ventana Medical Systems, Tucson, AZ, USA). A rabbit monoclonal p-MAPK (p44/p42; extracellular regulated kinase 1/2) antibody (Cell Signaling, Boston, MA, USA catalog no. 43707) was used at a 1 μg ml−1 concentration. The tissue sections were blocked for 30 min in 10% normal goat serum, 2% BSA in PBS. The incubation with the primary antibody was done for 5 h, followed by 60 min incubation with biotinylated goat anti-rabbit IgG (Vector Labs, Burlingame, CA, USA, cat no. PK6101) at a 1:200 dilution. DABMAP kit (Ventana Medical Systems) was used according to the manufacturer instructions. Digital images were obtained by the Mirax Scanner (Carl Zeiss, Dublin, CA, USA). Images were reviewed and confirmed by a breast pathologist at MSKCC.
Western blotting
Cell lysates pre and post infection were prepared at different time points and tested against various primary antibodies, including rabbit polyclonal anti-phospho p44/p42 MAPK antibody (Cell Signaling); rabbit polyclonal anti-phospho Akt antibody (Cell Signaling); rabbit polyclonal anti-phospho S6 antibody (Cell Signaling); rabbit polyclonal anti-p44/p42 MAPK antibody (Cell Signaling); rabbit polyclonal anti-MEK antibody (Cell Signaling); rabbit polyclonal anti-Akt antibody (Cell Signaling); rabbit polyclonal anti-S6 antibody (Cell Signaling); mouse polyclonal anti-infected cell protein (ICP) 8 and anti-ICP 27 antibody (Abcam, Cambridge, MA, USA); goat polyclonal anti-Actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Synergy study with MEK inhibitor PD98059
3 × 104 cells per well of HCC1937 were plated in 24-wells overnight. Drug A=NV1066, Drug B=PD98059 and combination A and B were added (using a constant ratio combination design) to each well and incubated for 4 days. Lactate dehydrogenase assay was used to determine each drug effect. The Chou and Talalay combination index (CI) method25, 26 was used to determine the effects of PD98059 on NV1066. Combination analysis was performed by using the CI isobologram analysis and the Composyn software program (Paramus, NJ, USA)27 for automated and dose-effect analysis. Using this method, CI values less than one indicate synergism, values of CI equal to one indicate additive effects and values of CI greater than one indicate antagonism. In addition, the Chou and Talalay algorithm was applied to the calculate dose-reduction index for each drug in their combination.
Ethics statement
The animal protocol no. 06-07-015 entitled ‘Oncolytic Viral Therapy’ was approved by the Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center. Last annual renewal was approved in January of 2012.
Results
NV1066 infects, replicates within and effectively kills TNBC by inducing cytolysis in vitro
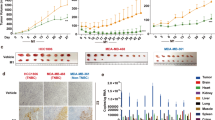
Viral infectivity and GFP expression were assessed by fluorescence microscopy 1 and 3 days after viral infection at an MOI of 0.1 and 1 (Figure 1a). GFP expression confirmed viral infection by 24 h in all cell lines and was time dependent, with maximum expression by day 3. Viral infection was also concentration dependent in all cell lines with increased expression at an MOI of 1 compared with MOI of 0.1.

NV1066 infects, replicates and demonstrates effective growth inhibition in multiple TNBC cell lines. (a) GFP expression confirmed viral infection as early as 24 h. Viral infectivity is dose dependent (shown in upper panel for day 1) and time dependent (shown at an MOI of 0.1 for days 1 and 3 in the lower panel). (b) Survival curves of infected cells compared with uninfected controls were calculated using a lactate dehydrogenase assay using different MOIs of NV1066 (0.01, 0.1, and 1). All cytotoxicity assays were carried out for 7 days in all five TNBC cell lines. MOI, Multiplicity of infection. (c) NV1066 replicates in all TNBC cell lines efficiently. Standard viral plaque assay was performed to determine the replication efficacy of NV1066 in five TNBC cell lines. Highest viral titer of 5 × 106 PFU, a 170-fold increase from initial viral dose at an MOI of 1.0 within 3 days of viral infection. Data represent means±s.e.m.
NV1066 reached near-complete cell kill in all TNBC cell lines effectively (Figure 1b). Greater than 90% cell kill was achieved in more-sensitive lines MDA-MB-231, HCC1806 and HCC38 by day 6 at the lowest MOI of 0.1. NV1066 still had a significant cytotoxic effects in the cell lines (HCC1937, HCC1143), achieving >70% cell kill by day 7 (MOI 1); however, these cell lines were less sensitive to viral therapy. Standard viral plaque assays were performed to assess viral replication in TNBC cell lines. After infection with NV1066 at an MOI of 1, all TNBC cell lines allowed efficient viral replication with the highest viral titer of 5 × 106 PFU, a 170-fold increase from initial viral dose at an MOI of 1 (Figure 1c).
NV1066 significantly downregulates MEK/MAPK pathway in infected TNBC in vitro and in vivo
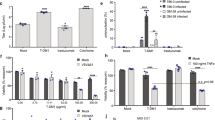
We evaluated the effects of NV1066 on activation of MAPK in all cell lines. Treatment of all the cell lines with NV1066 at an MOI of 1 for 24 h produced a significant reduction in MAPK phosphorylation (Figure 2a). Activation of MAPK was almost completely abolished following treatment with NV1066 after 48 h. Similar effects were seen in p-MEK levels. No significant changes were observed in Akt or S6 signaling pathway during viral replication (Figure 2b). When comparing baseline p-MAPK level among all TNBC cells lines, the more-sensitive cell lines (MDA-MB-231, HCC1806 and HCC38) were noted to have higher baseline p-MAPK levels compared with the less-sensitive lines (HCC1937, HCC1143) (Figure 2c).

(a) NV1066 downregulates the MEK/MAPK pathway in TNBC in vitro. Immunoblotting of MAPK pre and post viral treatment in five TNBC cell lines. Both phosphorylated and non-phosphorylated MAPK are shown. Actin is shown as loading control. Time of collection of cell lysates after infection is shown in hours. (b) NV1066 has no significant effects on PI3K and mTOR signaling pathway in TNBC in vitro. Immunoblotting of Akt and S6 pre and post viral treatment with NV1066 in 2 TNBC cell lines, HCC1837 and HCC38. Both phosphorylated and non-phosphorylated Akt and S6 are shown as components of PI3K/mTOR signaling pathway. Actin is shown as loading control. Time of collection of cell lysates after infection is shown in hours. (c) Baseline expression of p-MAPK in TNBC cell lines. Western blotting of phosphorylated and total MAPK expression from lysates from non-treated TNBC cell lines. Twenty microgram of protein was loaded per well. Cell lines MDA-MB-231, HCC1806 and HCC38 showed higher baseline expression of p-MAPK compared with HCC1937 and HCC1143. Higher expression level of p-MAPK correlated with increased sensitivity to viral oncolysis (indicated by increasing arrow sign). Actin antibody was used for equal loading.
NV1066 can effectively treat primary TNBC tumors in vivo
To confirm that the sensitivity of NV1066 is not specific to in vitro growth in monolayer culture, we sought in vivo confirmation of our cytotoxicity finding. We tested antitumor activity of a single injection of NV1066 (1 × 107 PFU in 100 μl) in flank xenografts of the cell line, MDA-MB-231, 2 weeks after tumor formation. Mean volume of flank tumors 2 weeks after treatment with NV1066 was 57 mm3 versus 438 mm3 in untreated controls (P=0.002) (Figure 3a). All mice tolerated viral therapy well. No difference in body weight was noted between the treatment and control group. In vivo, treated tumors showed decreased levels of p-MAPK on immunohistochemistry staining compared with control tumors (Figure 3b).

NV1066 treats TNBC xenografts efficiently and downregulates activation of MAPK signaling in vivo. (a) Flank tumors with MDA-MB-231 were treated with 100 μl of PBS (control) or 1 × 107 PFU of NV1066. Arrow indicates time of viral treatment (day 0). Data represent means±s.e.m. with n⩾6 per group. *P<0.05. (b) Representative images of p-MAPK immunostaining for cryo-frozen tumor tissues in MDA-MB-231 tumors. Images represent IHC staining of tumors after 2 weeks of treatment compared with controls. Treated sections demonstrate less staining for p-MAPK compared with control tumors.
Treatment with NV1066 and PD98059 synergistically inhibit the MEK/MAPK pathway
Previous research efforts suggest a model in which deactivation of p-MAPK via MEK inhibitor PD98059 has synergistic effects in combination with anti-EGFR or mTOR inhibitors. To evaluate the relative contribution of the MAPK pathway to viral sensitivity of NV1066 in TNBCs, we tested the effect of PD98059 in combination with NV1066 in the least sensitive cell line, HCC1937. Each compound alone had significant effect on cell viability; the combination of NV1066 and PD98059 resulted in a highly synergistic reduction in cell viability with a CI of <1.0. The synergistic effect manifested itself as a significantly lower combination EC50 as well as greater inhibition of cell viability across a wide range of concentrations (Table 1). Figure 4a demonstrates a Fraction-affected-Combination index (Fa-CI) plot created using the Chou and Talalay method and its CompuSyn software. All combination data points (all of which except for the very first point at low effect level/low concentration) fall below the CI=1 line, indicating synergism using a constant combination ratio. In our experiments, the calculated dose-reduction index for NV1066 and PD98059 demonstrated at least two- to sixfold for either single drug as compared with combination therapy (Table 2). Both MAPK and MEK were downregulated after infection with NV1066 depicted in Figure 4b. As expected by the synergistic antitumor effect of PD98059 and NV1066, and consistent with the observation of downregulation of p-MAPK in TNBC after infection with NV1066, combined therapy of PD98059 and NV1066 led to increased inhibition/downregulation of both p-MEK and p-MAPK compared with single therapy with NV1066 or PD98059. Furthermore, combination treatment led to increased levels of cleaved PARP, an indicator of apoptosis (Figure 4c).

NV1066 has synergistic effects with PD98059, both inhibiting MEK and downregulating the MAPK pathway. (a) Fraction-affected-Combination index (Fa-CI) plot using a constant ratio combination method. Plot was generated automatically by using CompuSyn. Combination index (CI) values<1 indicates synergism. (b) Immunoblotting of MEK and MAPK and (c) cleaved PARP (an indicator of apoptosis) from cell lysates used from the synergy experiment performed in 4A after 3 days of treatment. Actin is shown as loading control.
Discussion
TNBC is the one of the most aggressive breast tumors and is associated with dismal clinical outcomes. TNBC are unresponsive to standard hormonal or receptor-mediated therapies. Chemotherapy still remains the mainstay of therapy for TNBC. However, response rates are usually lower, and time to progression and metastatic spread is a common problem compared with other subtypes. Moreover, resistance can develop rapidly. Given these therapeutic shortcomings, novel targeted agents are needed to improve current clinical outcomes.
To our knowledge, this is the first report to show that oncolytic viral therapy uses HSV-1 as an effective therapeutic agent against TNBC. There are studies using vaccinia virus targeting on TNBCs.28, 29 In addition, our data demonstrate that the MEK/MAPK pathway is involved in the sensitivity of TNBC to NV1066. Indeed, our results exhibit that NV1066 can efficiently downregulate the activation of the MEK/MAPK pathway. Moreover, NV1066 proved to have synergistic effects with PD98059 in TNBC with the combination treatment resulting in increased suppression of tumor growth compared with single-drug therapy.
In any given tumor, there are multiple signaling pathways that have a role in regulating cell proliferation and differentiation. It has been shown that oncolytic viruses can make use of the activated signaling pathways of cancers.30, 31 This specific feature makes viral oncolytic therapy capable of targeting cancer cells while sparing normal cells. TNBC have been shown to express high levels of MEK/MAPK as compared with other pathways, such as the mTOR/PI3K signaling cascades.32, 33, 34, 35 Consistent with those results, our studies showed that NV1066 has a major effect on the MEK/MAPK pathway during viral infection; however, there were no significant effects on mTOR/PI3K signaling. NV1066 contains a deletion of the gene γ134.5 that specifically targets cancer cells with high MEK/MAPK expression and has been previously shown that the activity of the MEK signaling pathway in cancer cells is indicative of the susceptibility of the cell to γ134.5-deficient HSV cytotoxicity. Such HSVs have been shown to exhibit improved replication and, therefore, enhanced cytotoxicity in target cells with an activated MAPK pathway. For example, Veerapong et al.22 reported that systemic delivery of a γ134.5-deleted HSV-1 selectively targeted and killed human xenograft tumors that overexpress MEK activity compared with tumors that express lower MEK activity. In another study, Smith et al.36 tested multiple cell lines for sensitivity to a γ134.5-deleted HSV-1 and concluded that viral sensitivity hinged on endogenous kinase activity of the MEK protein, measured by the amount of extracellular regulated kinase 1/2 phosphorylation at the Thr202/Tyr204. Consistent with those results, in our study, cell lines with a higher levels of p-MAPK (MDA-MB-231, HCC1806, HCC38) were more sensitive to viral therapy compared with the ones with lower levels (HCC1937 and HCC1143). These preclinical findings also have potential utility in the clinical development of viral therapy because they suggest that tumor-specific MAPK activation could serve as a biomarker and facilitate clinical decision-making, specifically in patient selection for HSV viral therapy.
An important finding in our study was that viral therapy with NV1066 caused a significant decrease in p-MEK and p-MAPK without any significant changes in the mTOR/PI3K signaling pathways. This suggests that in these TNBC cells, the MAPK signaling pathway seems to be the main driver for proliferation and survival. Congruent with these results, our study demonstrates that NV1066 has synergistic effects with the MEK inhibitor, PD98059, in TNBC, signifying that inhibiting MEK/MAPK signaling with 2 agents in these cells has even greater effects on survival as compared with single therapy. The use of multiple drugs that act synergistically offers several benefits, such as increasing the therapeutic efficacy of each drug or decreasing the dosage but increasing or maintaining the same efficacy to avoid toxicity. Moreover, synergism could also help minimize the development of drug resistance.25 With regard to using dual-therapy in breast cancer, recent studies including the use of anti-human HER-2 monoclonal antibodies pertuzumab and trastuzumab have shown that dual-agent therapy is superior to monotherapy.37, 38, 39 In our combination experiments, the dose-reducing index curves calculated for NV1066 and PD98059 showed a dose-reducing index of at least two- to sixfold for either single drug as compared with combination therapy calculated by Chou–Talalay method. Moreover, as the cells continue to show high activation of S6 and Akt after viral infection, one could potentially combine NV066 with other signal protein inhibitors, such as PI3K inhibitor or and mTOR inhibitor, essentially targeting multiple pathways to achieve better therapeutic effects. Here, we report a potential target and treatment which merit further investigation.
Overexpression and activation of MAPK contribute to the resistance of TNBC to multiple therapies. Eralp et al.34 showed that p-MAPK expression was a significant prognostic factor in predicting recurrence in patients with TNBC. Anthracycline resistance and survival following relapse was significantly worse for patients with higher MAPK score. Other researchers have shown that increased expression of p-MAPK was associated with resistance to anti-EGFR agents such as gefitinib,32 radiation therapy,20 or the mTOR inhibitor rapamycin.35 Normanno et al.33 reported that gefitinib and PD98059 produced synergistic antitumor effect and increased apoptosis compared with single-agent treatment. The synergistic effect was associated with a profound decrease in MAPK activation. Another study by Zhao et al.35 showed that high doses of an mTOR inhibitor, CCI-779, had no effects on brain metastasis. But when MEK inhibitor (SL327) was combined with CCI-779, there was significant reduction of brain metastasis in vivo. Altogether, our results show that NV1066 downregulates the MEK/MAPK signaling pathway in TNBC in vitro and in vivo. We believe that NV1066 not only has cytotoxic effects but may also have a role in sensitizing TNBCs to other therapeutic agents, such as mTOR inhibitors for clinical application.
In summary, our results show that NV1066 can effectively suppress TNBC growth in vitro and in vivo. Furthermore, this study confirms the direct involvement of the MAPK signaling cascade in the sensitivity of TNBC cells to HSV-1 oncolytic viral therapy and demonstrated synergistic effects of NV1066 and the MEK inhibitor PD98059. The complete mechanism of the effects of this virus on the MAPK pathway is still unknown and merits further investigation. These findings provide a promising basis for the continued investigation of this virus as a novel therapeutic and sensitizing agent for treatment of this aggressive disease.
Abbreviations
- TNBCs:
-
triple-negative breast cancers
- MOI:
-
multiplicity of infection
- EGFR:
-
epidermal growth factor receptor
- MAPK:
-
mitogen-activated protein kinase
- mTOR:
-
mammalian target of rapamycin
- ERK:
-
extracellular regulated kinase
- HSV-1:
-
herpes simplex virus-1
- GFP:
-
green fluorescent protein
- PFU:
-
plaque-forming units
- LDH:
-
lactate dehydrogenase
- IACUC:
-
Institutional Animal Care and Use Committee
- MSKCC:
-
Memorial Sloan-Kettering Cancer Center
- CI:
-
combination index
- Fa-CI:
-
fraction-affected-combination index
- DRI:
-
dose-reducing index
References
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13 (15 Pt 1): 4429–4434.
Foulkes WD, Smith IE, Reis-Filho JS . Triple-negative breast cancer. N Eng J Med 2010; 363: 1938–1948.
Korsching E, Packeisen J, Agelopoulos K, Eisenacher M, Voss R, Isola J et al. Cytogenetic alterations and cytokeratin expression patterns in breast cancer: integrating a new model of breast differentiation into cytogenetic pathways of breast carcinogenesis. Lab Invest 2002; 82: 1525–1533.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 2004; 10: 5367–5374.
Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst. 2003; 95: 1482–1485.
Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. JSteroid Biochem Mol Biol 2002; 80: 239–256.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–279.
Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE . Oncolytic viruses in cancer therapy. Cancer Lett 2007; 254: 178–216.
Jacobs L . Positive margins: the challenge continues for breast surgeons. Ann Surg Oncol 2008; 15: 1271–1272.
Adusumilli PS, Gholami S, Chun YS, Mullerad M, Chan MK, Yu Z et al. Fluorescence-assisted cytological testing (FACT): Ex Vivo viral method for enhancing detection of rare cancer cells in body fluids. Mol Med 2011; 17: 628–634.
Carson J, Haddad D, Bressman M, Fong Y . Oncolytic Herpes Simplex Virus 1 (Hsv-1) Vectors: Increasing Treatment Efficacy and Range through Strategic Virus Design. Drugs Future 2010; 35: 183–195.
Kemeny N, Brown K, Covey A, Kim T, Bhargava A, Brody L et al. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther 2006; 17: 1214–1224.
Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 2000; 7: 867–874.
Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clinical Cancer Res 2010; 16: 4005–4015.
Kaufman HL, Bines SD . OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol 2010; 6: 941–949.
Adusumilli PS, Chan MK, Hezel M, Yu Z, Stiles BM, Chou TC et al. Radiation-induced cellular DNA damage repair response enhances viral gene therapy efficacy in the treatment of malignant pleural mesothelioma. Ann Surg Oncol 2007; 14: 258–269.
McAuliffe PF, Jarnagin WR, Johnson P, Delman KA, Federoff H, Fong Y . Effective treatment of pancreatic tumors with two multimutated herpes simplex oncolytic viruses. J Gastrointest Surg 2000; 4: 580–588.
Eisenberg DP, Carpenter SG, Adusumilli PS, Chan MK, Hendershott KJ, Yu Z et al. Hyperthermia potentiates oncolytic herpes viral killing of pancreatic cancer through a heat shock protein pathway. Surgery 2010; 148: 325–334.
Sivendran S, Pan M, Kaufman HL, Saenger Y . Herpes simplex virus oncolytic vaccine therapy in melanoma. Expert Opin Biol Ther 2010; 10: 1145–1153.
Stiles BM, Adusumilli PS, Stanziale SF, Eisenberg DP, Bhargava A, Kim TH et al. Estrogen enhances the efficacy of an oncolytic HSV-1 mutant in the treatment of estrogen receptor-positive breast cancer. IntJ Oncol 2006; 28: 1429–1439.
Sze DY, Iagaru AH, Gambhir SS, De Haan HA, Reid TR . Response to intra-arterial oncolytic virotherapy with the herpes virus NV1020 evaluated by [18F]fluorodeoxyglucose positron emission tomography and computed tomography. Hum Gene Ther 2012; 23: 91–97.
Veerapong J, Bickenbach KA, Shao MY, Smith KD, Posner MC, Roizman B et al. Systemic delivery of (gamma1)34.5-deleted herpes simplex virus-1 selectively targets and treats distant human xenograft tumors that express high MEK activity. Cancer Res 2007; 67: 8301–8306.
Wong RJ, Joe JK, Kim SH, Shah JP, Horsburgh B, Fong Y . Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Hum Gene Ther 2002; 13: 1213–1223.
Adusumilli PS, Eisenberg DP, Chun YS, Ryu KW, Ben-Porat L, Hendershott KJ et al. Virally directed fluorescent imaging improves diagnostic sensitivity in the detection of minimal residual disease after potentially curative cytoreductive surgery. J Gastrointest Surg 2005; 9: 1138–1146 discussion 46-7.
Chou TC . Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–681.
Chou TC, Talalay P . Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55.
Chou TC, Martin N CompuSyn for drug combinations: PC software and user's guide, 2005. Available from http://www.combosyn.com/index.html.
Gil M, Seshadri M, Komorowski MP, Abrams SI, Kozbor D . Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc Natl Acad Sci oUSA 2013; 110: E1291–E1300.
Gholami S, Chen CH, Lou E, De Brot M, Fujisawa S, Chen NG et al. Vaccinia virus GLV-1h153 is effective in treating and preventing metastatic triple-negative breast cancer. Ann Surg 2012; 256: 437–445.
Farassati F, Yang AD, Lee PW . Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol 2001; 3: 745–750.
Guo ZS, Thorne SH, Bartlett DL . Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta 2008; 1785: 217–231.
Normanno N, Campiglio M, Maiello MR, De Luca A, Mancino M, Gallo M et al. Breast cancer cells with acquired resistance to the EGFR tyrosine kinase inhibitor gefitinib show persistent activation of MAPK signaling. Breast Cancer Res Treatment 2008; 112: 25–33.
Normanno N, De Luca A, Maiello MR, Campiglio M, Napolitano M, Mancino M et al. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol 2006; 207: 420–427.
Eralp Y, Derin D, Ozluk Y, Yavuz E, Guney N, Saip P et al. MAPK overexpression is associated with anthracycline resistance and increased risk for recurrence in patients with triple-negative breast cancer. Ann Oncol 2008; 19: 669–674.
Zhao H, Cui K, Nie F, Wang L, Brandl MB, Jin G et al. The effect of mTOR inhibition alone or combined with MEK inhibitors on brain metastasis: an in vivo analysis in triple-negative breast cancer models. Breast Cancer Res Treat 2012; 131: 425–436.
Smith KD, Mezhir JJ, Bickenbach K, Veerapong J, Charron J, Posner MC et al. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1. J Virol 2006; 80: 1110–1120.
Revannasiddaiah S, Seam R, Gupta M . Pertuzumab plus trastuzumab in metastatic breast cancer. N Engl J Med 2012; 366: 1349 (author reply 50).
Bender E . Dual HER2 blockade slows metastatic breast cancer. Cancer Discov 2012; 2: OF4.
Guarneri V, Frassoldati A, Bottini A, Cagossi K, Bisagni G, Sarti S et al. Preoperative chemotherapy plus trastuzumab, lapatinib, or both in human epidermal growth factor receptor 2-positive operable breast cancer: results of the randomized phase II CHER-LOB study. J Clin Oncol 2012; 30: 1989–1995.
Acknowledgements
Technical services provided by the Research Animal Resource Center and the Molecular Cytology at Memorial Sloan-Kettering Cancer Center are gratefully acknowledged. This project was supported by the Flight Attendant Research Fund.
Author Contributions
SG and CC designed the entire study together, carried out all major experiments with the help of others and primarily drafted the manuscript. SG contributed to the MEK inhibitor study. SG and EL helped with the design of the study and helped write the manuscript. SF assisted with immunohistochemistry study. JC contributed to the cytotoxicity assay. JN and TC participated and contributed to the drug combination experiment. JB and YF contributed to the study design and complete of the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Gholami, S., Chen, CH., Gao, S. et al. Role of MAPK in oncolytic herpes viral therapy in triple-negative breast cancer. Cancer Gene Ther 21, 283–289 (2014). https://doi.org/10.1038/cgt.2014.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2014.28
This article is cited by
-
Oncolytic HSV1 targets different growth phases of breast cancer leptomeningeal metastases
Cancer Gene Therapy (2023)
-
Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy
Molecular Cancer (2020)
-
Integrating oncolytic viruses in combination cancer immunotherapy
Nature Reviews Immunology (2018)
-
Oncolytic virotherapy for urological cancers
Nature Reviews Urology (2016)
-
HSV-1 as a novel therapy for breast cancer meningeal metastases
Cancer Gene Therapy (2015)
