
Abstract
Targeted oncolytic poxviruses hold promise for the treatment of cancer. Arming these agents with immunostimulatory cytokines (for example, granulocyte-monocyte colony-stimulating factor; GM-CSF) can potentially increase their efficacy and/or alter their safety. However, due to species-specific differences in both human GM-CSF (hGM-CSF) activity and poxviruses immune avoidance proteins, the impact of hGM-CSF expression from an oncolytic poxvirus cannot be adequately assessed in murine or rat tumor models. We developed a rabbit tumor model to assess toxicology, pharmacodynamics, oncolytic efficacy and tumor-specific immunity of hGM-CSF expressed from a targeted oncolytic poxvirus JX-963. Recombinant purified hGM-CSF protein stimulated a leukocyte response in this model that paralleled effects of the protein in humans. JX-963 replication and targeting was highly tumor-selective after i.v. administration, and intratumoral replication led to recurrent, delayed systemic viremia. Likewise, hGM-CSF was expressed and released into the blood during JX-963 replication in tumors, but not in tumor-free animals. hGM-CSF expression from JX-963 was associated with significant increases in neutrophil, monocyte and basophil concentrations in the peripheral blood. Finally, tumor-specific cytotoxic T lymphocytes (CTL) were induced by the oncolytic poxvirus, and expression of hGM-CSF from the virus enhanced both tumor-specific CTL and antitumoral efficacy. JX-963 had significant efficacy against both the primary liver tumor as well as metastases; no significant organ toxicity was noted. This model holds promise for the evaluation of immunostimulatory transgene-armed oncolytic poxviruses, and potentially other viral species.
Similar content being viewed by others
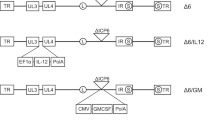

Introduction
Oncolytic viruses (virotherapy) kill cancer cells through a novel mechanism-of-action; oncolysis has features of both necrosis and apoptosis. Oncolytic virus replication can be targeted to cancer cells specifically. Selective intratumoral replication leads to virus multiplication, lysis of the infected cancer cell and spread to adjacent and distant cancer cells. In addition, these therapeutics can kill through additional mechanisms including induction of tumor-specific cytotoxic T lymphocytes (CTL), and ‘arming’ for expression of therapeutic transgene products. Therefore, armed virotherapeutics have multiple attractive features needed in a novel cancer treatment platform.
Granulocyte-monocyte colony-stimulating factor (GM-CSF) has shown great promise in strategies to induce tumor-specific CTLs.1 For this reason, the human GM-CSF gene (hGM-CSF) has been engineered into several oncolytic viruses including adenoviruses,2 herpes viruses3 and vaccinia.4 Murine GM-CSF (mGM-CSF) expression increased the injection site efficacy of a replication-selective oncolytic adenovirus in immunodeficient mice following intratumoral injection.5 An attenuated oncolytic herpes virus (HSV) expressing murine GM-CSF (mGM-CSF) demonstrated tumor growth inhibition in mice at distant sites following intratumoral injection; distant efficacy was superior to that with a mGM-CSF− control virus. A hGM-CSF-expressing vaccinia virus (JX-594) demonstrated objective tumor responses following intratumoral injection in patients with refractory cancers in the liver; responses were demonstrated at both injection sites and in distant, noninjected tumors.6, 7 JX-594 replicated, spread and was released into the blood resulting in infection of distant noninjected tumor sites; T-cell infiltrations and regressions of distant skin metastases have also been demonstrated.6, 8 Similar findings were noted with another oncolytic poxvirus, JX-963, in preclinical models.9 In summary, hGM-CSF-expressing oncolytic viruses hold potential for cancer treatment.
A major hurdle for the development of these promising therapeutics, however, is the lack of a validated animal tumor model to assess the effects of hGM-CSF expression from oncolytic poxviruses. For example, one problem is that murine and hGM-CSF proteins have important differences in their activities. mGM-CSF induces only GM colonies, whereas hGM-CSF induces various types of colonies including GM, eosinophil, mast cell, erythrocyte, megakaryocyte, blast cell and mixed hemopoietic colonies.10 In addition, hGM-CSF itself is not fully active in mice or rats.11, 12 Finally, a number of poxvirus immune avoidance gene products are partially or completely inactive in mice. We therefore sought to develop an immunocompetent tumor model that had the following features: (1) hGM-CSF had similar activity to that shown in humans, (2) vaccinia replicated efficiently and (3) vaccinia immune avoidance proteins were active. For these studies we tested matched targeted vaccinia viruses with hGM-CSF expression (JX-963) and without it (vvDD).
Result
Characterization of vvDD (without GM-CSF) and JX-963 (with GM-CSF) in vitro
The viruses in this study were generated from the Western Reserve vaccinia virus-derived vvDD backbone with deletions in both the thymidine kinase gene and the vaccinia growth factor (vgf) gene; these two deletions resulted in a significant increase in safety and cancer selectivity.13 The vvDD expressing hGM-CSF under control of the synthetic early-late promoter was designated JX-963.7, 9 As was described previously,2, 7, 9 JX-963 was constructed from vvDD by insertion of hGM-CSF into the TK gene (Supplemental Figure 1). The GM-CSF production from a panel of four cancer cells was compared in vitro after infection with vvDD, JX-963 or JX-594 infection (control vaccinia expressing GM-CSF; GM-CSF inserted into Wyeth strain vaccinia into disrupted TK region; Supplemental Table 1).6 As was expected, no GM-CSF was produced by vvDD, whereas both JX-963 and JX-594 produced GM-CSF from all cells at 24 h postinfection. The in vitro replication rates of vvDD and JX-963 showed no significant difference (Supplemental Figure 2); JX-963 replication was significantly higher than that with JX-594 (approximately 10-fold higher at 72 h; P<0.05).
Development and characterization of a novel VX2 rabbit liver cancer model for use with hGM-CSF-expressing oncolytic poxviruses
An immunocompetent, orthotopic and spontaneously metastasizing rabbit (New Zealand White; NZW) VX2 tumor model was established as follows. VX2 tumor fragments (1 mm3) were implanted into hepatic parenchyma and the implantation site was subsequently covered with Surgicel patch, which prevented initial dissemination of tumor cells. Both primary tumor growth and systemic metastases development and growth were monitored by serial weekly CT scans. Reproducible time-dependent development of multiple lung and liver metastases (contralateral lobe) was demonstrated (Figure 1a–d).

VX2 intrahepatic tumor in inbred New Zealand Rabbit model. (a) Time dependent increase of VX2 hepatic mass determined by CT. (b) Time dependent increase of pulmonary metastasis determined by CT. (c) Representative CT of primary hepatic mass (left, red arrow) and lung metastasis (right, red arrow). (d) Representative gross image of primary hepatic mass (left) and histology of metastatic pulmonary nodule (right).
Pharmacokinetics and pharmacodynamics of recombinant human GM-CSF protein in the NZW rabbit
The inbred rabbit was selected in this study as the in vivo test species for efficacy and safety testing of JX-963 based on literature references indicating that hGM-CSF has biological activity in that species, as well as extensive literature citations on the use of the rabbit for vaccinia research.13 However, there is no direct evidence on hematological effect of recombinant human GM-CSF protein (rhGM-CSF) in rabbit species. Therefore, in response to a request from US FDA, we elected to determine the effects of daily dosing with hGM-CSF protein, at doses similar to those used in humans, on subpopulations of WBC in the normal rabbit.
Serial hematological changes were observed after s.c. injection (daily for 10 days, 40 μg kg−1) of rhGM-CSF in these normal rabbits to determine hGM-CSF activity. rhGM-CSF significantly increased the total WBC on days 2, 4, 6 and 11 (Figure 2a). The transient decrease in total WBC in the control group was presumably due to daily blood sampling. On the WBC differential analysis, absolute neutrophil, monocyte and basophil concentrations were significantly increased (Figure 2b–d). Plasma concentrations of hGM-CSF in samples taken immediately before treatment (trough levels) on each of the blood sampling days ranged from 12.3 to 37.8 pg ml−1 (Supplemental Figure 3) indicating that prolonged exposure (at least 24 h) to hGM-CSF occurred as a consequence of the daily subcutaneous administration. These concentrations are similar to concentrations in humans treated with rhGM-CSF. To our knowledge, these results are the first report of the direct effect of hGM-CSF protein on WBC populations in the rabbit or any rodent. These results confirm similar pharmacodynamic effects of hGM-CSF in NZW rabbits and humans.

Hematological effects of recombinant human granulocyte-monocyte colony-stimulating factor (hGM-CSF) protein in normal New Zealand White (NZW) rabbits. The effect of serial rhGM-CSF protein s.c. on days 1–10 was studied including effects on blood concentrations of (a) total WBC, (b) neutrophils, (c) monocytes, (d) basophils (* statistically significant difference vs control group).
Biodistribution and pharmacodynamics of vvDD and JX-963 after intravenous injection in VX2 rabbit tumor model
vvDD (no hGM-CSF) or JX-963 (vvDD encoding hGM-CSF) were administered i.v. to rabbits with VX2 primary liver tumors and systemic metastases. The biodistribution of vvDD and JX-963 were assessed at serial time points (four rabbits per time point: two male, two female). Tumor and normal organ tissue samples were harvested and viral titers were assessed as described. Significant increases and persistence of viral titers over time, indicative of replication, were observed in tumor tissues on days 1−22 after i.v. administration of vvDD or JX-963 (Figure 3a). Serum hGM-CSF was measured in normal, tumor-free rabbits or in the VX2-bearing rabbit model after i.v. injection of vvDD or JX-963. As shown in Figure 3b, measurable hGM-CSF was detected in serum obtained from VX2-bearing rabbits treated with JX-963 (hGM-CSF expressing). However, hGM-CSF was detected only at extremely low concentrations, or was undetectable, in serum after i.v. administration in tumor-free rabbits (Figure 3a) suggesting i.v. administration, JX-963 replication and hGM-CSF production was highly tumor-specific. As expected, vvDD did not lead to detectable serum GM-CSF at doses even 10-fold higher than that with JX-963 (data not shown).

Tumor-specific replication, biodistribution and serum granulocyte-monocyte colony-stimulating factor (GM-CSF) protein expression after i.v. administration of JX-963 in the VX2 liver tumor model. (a) Biodistribution of vvDD and JX-963 in tumors vs normal organs over time following intravenous injection. (b) hGM-CSF serum concentrations over time following intravenous injection of JX-963 in tumor-free and VX2-bearing rabbits.
Immune response parameters after intravenous injection of vvDD and JX-963: induction of neutralizing antibodies and tumor-targeting cytotoxic T lymphocytes
To assess the immune response against both the product itself (neutralizing antibodies) and against the target tumor (tumor-specific CTL), we evaluated these parameters after i.v. vvDD or JX-963. Neutralizing antibodies to JX-963 became detectable as soon as 5 days after i.v. injection and rose through day 18 (Supplemental Figure 4); a dose-response trend was notable (P<0.05 at days 8 and 18). We subsequently compared neutralizing antibody titers in day 18 serum after i.v. JX-963 or vvDD. Serum antibody titers were not significantly different after JX-963 or vvDD injection (data not shown).
For CTL assays against VX2 tumor cells, on in vitro target cell assay had to be developed. VX2 tissues are typically maintained by serial in vivo passage by intramuscular injection in rabbits; to our knowledge, VX2 cell lines have not been derived and perpetuated for in vitro passage. We generated VX2 single cell suspensions by enzymatic treatment as described in the methods section, and after eight passages cells were used as in vitro target cells. Splenocytes were isolated from VX2-bearing rabbits before and after multiple injections of i.v. phosphate-buffered saline (PBS), vvDD or JX-963. vvDD injection led to a significant increase in VX2-targeting CTL vs PBS as predicted (Figure 4a; P< 0.05). In addition, JX-963 CTL induction was significantly higher than with vvDD, presumably due to hGM-CSF expression within the tumor milieu (Figure 4b; P< 0.05).

Immune Response parameters after i.v. injection of vvDD (GM-CSF−) or JX-963 (GM-CSF+). (a) Antivaccinia neutralizing antibody development over time. (b) Induction of VX2 tumor-targeting cytotoxic T lymphocytes (CTL) following i.v. administration of vvDD or JX-963 (*, P<0.05 between groups); E/T ratio, ratio of effector cells to target cells.
Efficacy of vvDD and JX-963 against primary and metastatic tumors following intravenous administration
Antitumoral efficacy against primary and metastatic tumor foci was evaluated by serial chest and abdominal CT scanning in this VX2 tumor model (see above). As previously described, vvDD had significant efficacy in the VX2 model at a dose of 3 × 108 p.f.u. kg−1 i.v.4 To determine whether the expression of hGM-CSF from vvDD (JX-963) led to improved efficacy, we administered animals with a subtherapeutic dose of i.v. vvDD (3 × 107 p.f.u. kg−1). Of note, diffuse microscopic metastases were present in control animals killed at the same time as treatment was started; these metastases were not detectable by CT at the time. Both vvDD and JX-963 had efficacy against the large established primary tumor within the liver (Figure 5a; P<0.05 vs PBS); by week 8, JX-963 efficacy was significantly greater than that with vvDD. Both vvDD and JX-963 were effective at preventing the outgrowth of CT-detectable tumor metastases in liver and lung (Figure 4b; P<0.05 vs PBS). JX-963 was able to completely prevent the outgrowth of detectable lung or liver metastases in these animals (Figure 5b; P<0.05 vs vvDD). Rabbits treated with PBS or this relatively low dose of vvDD had a median survival of 7 or 9 weeks, respectively, vs 14 weeks in the JX-963 injected rabbit group (Figure 5c; P<0.05). JX-963-treated rabbits also gained weight whereas PBS treated animals lost weight (data not shown). Therefore, the vvDD oncolytic vaccinia virus had systemic efficacy against both large established primary liver tumors and systemic micrometastases, and arming with hGM-CSF further increased systemic anticancer efficacy and survival.

Antitumoral efficacy of i.v. vvDD and JX-963 in VX2 tumor model. (a) Growth curves of primary hepatic tumors in groups treated i.v. with vvDD, JX-963 or phosphate-buffered saline (PBS; control). (b) Antitumoral efficacy against lung metastases (week 8 after tumor implantation) for groups treated i.v. with vvDD, JX-963 or PBS (control). (c) Kaplan–Meier survival curves for overall survival of treatment groups.
GLP toxicology study of i.v. JX-963 in NZW rabbits
To prepare for a phase I clinical trial with JX-963, we performed a toxicity study with i.v. JX-963 under Good Laboratory Practice (GLP) guidelines. Doses were 3 × 107 p.f.u. kg−1 and 1 × 108 p.f.u. kg−1 as either once or thrice weekly doses. In a subsequent non-GLP study, a dose of 1 × 109 p.f.u. kg−1 in the same treatment regimens was evaluated. These doses were up to 30-fold higher than efficacious doses in tumor-bearing animals. No animal deaths were reported after either single-dose or multiple-dose i.v. injection. Only at the highest dose level, transient rapid breathing and inactivity was noted postdosing but resolved within 1 h; lower dose animals had no change in activity. Mild-dose-related decreases in body weights were noted following dosing, but weight gain resumed after 2 weeks in all cases. Changes in hematology included transient monocytosis and thrombocytosis in treated animals. No normal tissue toxicities were evident on histopathology analyses even though virus replication-based quantitative PCR was observed transiently in some normal tissues for 1–5 days. The only treatment-related histopathology findings consisted of perivascular monocytic inflammation at injection sites (data not shown).
Discussion
Cody et al.14 reported that dendritic cells could be generated in vitro from rabbit bone marrow cells using a combination of hGM-CSF, interleukin-4 and LPS, indicating cross-species reactivity. Human GM-CSF given intraperitoneally to neonatal rabbits increased blood eosinophil and granulocyte counts, an effect similar to that reported after administration of hGM-CSF to humans.15 Taken together, these papers indicate that hGM-CSF has cross-species reactivities in the rabbit that are qualitatively similar to some of those reported in the human for this multilineage hematopoietin.16 However, neither paper provided quantitative data on the effects of hGM-CSF on populations of WBC in the rabbit that could be compared with those reported for the human.
We sought to develop an immunocompetent tumor model that had the following features: (1) hGM-CSF had similar activity to that shown in humans, (2) vaccinia replicated efficiently and (3) vaccinia immune avoidance proteins were active. As poxviruses replicate efficiently in NZW rabbits, and many immunomodulatory poxvirus proteins are active in rabbits, we elected to develop an orthotopic, immunocompetent rabbit tumor model. For these studies we tested matched targeted vaccinia viruses with hGM-CSF expression (JX-963) and without it (vvDD). In this study, for the first time to our knowledge, we demonstrated direct hematologic effects of hGM-CSF protein in rabbits that parallel effects in humans. Significant increases were noted in neutrophils and monocytes. vvDD and JX-963 replication and targeting were highly tumor-selective after i.v. administration, and intratumoral replication led to recurrent, delayed systemic viremia. The reason for initial accumulation of JX-963 into tumor environment after i.v. administration may be due to leaky tumor microvasculature as we have observed high dose of virus in tumor tissues after either vvDD or JX-963 administration.
Likewise, hGM-CSF was expressed and released into the blood during JX-963 replication in tumors, but not in tumor-free animals. hGM-CSF expression from JX-963 was associated with significant increases in neutrophil, monocyte and basophil concentrations in the peripheral blood. Finally, tumor-specific CTL were induced by the oncolytic poxvirus, and expression of hGM-CSF from the virus enhanced both tumor-specific CTL and antitumoral efficacy. JX-963 had significant efficacy against both the primary liver tumor as well as metastases; no significant organ toxicity was noted. This model holds promise for the evaluation of immunostimulatory transgene-armed oncolytic poxviruses, and potentially other viral species. Animal tumor models with better predictive value should lead to improved optimization of these products, and as a result better clinical benefits for patients.
Methods
Viruses
The construction of vvDD was described previously;13 vvDD is a TK− and VGF− engineered Western Reserve strain vaccinia virus. JX-963, which expresses hGM-CSF from the disrupted TK gene region under control of the synthetic early-late promoter, was described previously.6
Rabbit VX2 tumor model studies
VX2 tumors were grown in the muscle of inbred NZW rabbits (Samtako, Oh-San, Korea) and cells from a 1–2 mm3 fragment of tumor were dissociated, resuspended in 0.1 ml normal saline and injected via a 21-gauge needle beneath the liver capsule. The injection site was covered with a Surgicel patch followed by a purse-string tie to prevent release of VX2 cells from the injection site.
Cytotoxic T-lymphocyte study
Isolated VX2 cells were used for target cells, and splenocytes isolated from VX2-bearing rabbits after JX-963, vvDD or PPS multiple injection ( × 3) were used for effector cells. VX2 cell lines were isolated from VX2 tissues enzymatically (collagenase 0.01 %+proteas 0.1 % O/N at 4 C), and maintained in vitro with Dulbecco's modified Eagle's medium with 10 % fetal bovine serum (FBS) for 8 passages. However VX2 cells can not be grown after storage at −80 °C, and we could not keep this study under more controlled environment. For isolation of splenocytes, spleens were harvested at the time of killing (1 week after i.v. dosing weekly for 3 weeks) and fractionated through a 70-mm nylon cell strainer. After red blood cell lysis, the splenocyte pool was suspended in RPMI 1640 medium with 90 % FBS, and then stored at −80 °C before CTL measurement. CTL activity was measured by fluorescence-activated cell sorting using the ACT1 assay (Cell Technology, Mountain View, CA).
Effects of hGM-CSF in rabbit model
Leukokine (hGM-CSF recombinant protein; LG Lifescience, Seoul, Korea) or control solution was administered s.c. daily for 10 days (days 1–10) to rabbits.
Measurement of serum antibodies to JX-963 or vvDD
The titer of antivaccinia NAb was determined by measuring the ability of the serum to protect A2780 cells from death caused by viral infection. Diluted serum samples were incubated with JX-963 or vvDD for 2 h, and the mixture was inoculated onto an A2780 cell monolayer. Cell viability was measured 3 days after inoculation by means of colorimetric assay (CCK-8; Dojindo, Inc., Kumamoto, Japan).
Statistical analyses
Kaplan–Meier curves were compared using the generalized Wilcoxin test. Tumor response rates and metastasis-free rates were compared with Fisher's exact test.
Live rabbits (3 rabbits among 5) on week 14 were simply killed at this point. Tumor size was monitored by CT every 1–2 weeks. CT screening at 10 week was not included as we did not anticipate that JX-963-injected rabbits can live for more than 14 weeks. The number of animals are different between groups; four rabbits in each control and vvDD group and five rabbits in JX-963 group. We included many control data as we did a lot of control vx2 model in a separate study. In most cases done in different time, control rabbits died within 5–9 weeks without significant deviation.
Conflict of interest
Except Dr David Kirn and Tae-Ho Hwang, all authors declare no conflict of interest. Dr David Kirn is a founder and CEO of Jennerex Biotechnology Company, which has been developing JX-963. Dr Tae-Ho Hwang has consulted for Pharmaceuticals and received compensation.
References
Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA 1993; 90: 3539–3543.
Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, Ganesh S et al. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor—armed oncolytic adenovirus for the treatment of bladder cancer. Clin Cancer Res 2006; 12: 305–313.
Kohno SI, Luo C, Nawa A, Fujimoto Y, Watanabe D, Goshima F et al. Oncolytic virotherapy with an HSV amplicon vector expressing granulocyte-macrophage colony-stimulating factor using the replication-competent HSV type 1 mutant HF10 as a helper virus. Cancer Gene Ther 2007; 14: 918–926.
Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther 2006; 14: 361–370.
Malhotra S, Kim T, Zager J, Bennett J, Ebright M, D'Angelica M et al. Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery 2007; 141: 520–529.
Park BH, Hwang TH, Liu TC, Sze DY, Kim JS, Kwon HC et al. Phase I clinical proof-of-concept trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with refractory primary or metastatic liver cancers. Lancet Oncol 2008; 9: 5330–5342.
Liu TC, Hwang TH, Park BH, Bell J, Kirn DH . The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 2008; 16: 1637–1642.
Mastrangelo MJ, Maguire Jr HC, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther 1999; 6: 409–422.
Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest 2007; 117: 3350–3358.
Nishijima I, Nakahata T, Hirabayashi Y, Inoue T, Kurata H, Miyajima A et al. A human GM-CSF receptor expressed in transgenic mice stimulates proliferation and differentiation of hemopoietic progenitors to all lineages in response to human GM-CSF. Mol Biol Cell 1995; 6: 497–508.
Murray HW . Effect of granulocyte–macrophage colony-stimulating factor in experimental visceral leishmaniasis. J Clin Invest 1995; 95: 1183–1192.
Shanafelt AB, Johnson KE, Kastelein RA . Identification of critical amino acid residues in human and mouse granulocyte–macrophage colony-stimulating factor and their involvement in species specificity. J Biol Chem 1991; 266: 13804–13810.
Isaacs SN, Kotwal GJ, Moss B . Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Proc Natl Acad Sci USA 1992; 89: 628–632.
Cody V, Shen H, Shlyankevich M, Tigelaar RE, Brandsma JL, Hanlon DJ . Generation of dendritic cells from rabbit bone marrow mononuclear cell cultures supplemented with hGM-CSF and hIL-4. Vet Immunol Immunopathol 2005; 103: 163–172.
Demetri GD, Antman KH . Granulocyte-macrophage colony-stimulating factor (GM-CSF): preclinical and clinical investigations. Semin Oncol 1992; 19: 362–385.
Sieff CA, Niemeyer CM, Nathan DG, Ekern SC, Bieber FR, Yang YC et al. Stimulation of human hematopoietic colony formation by recombinant gibbon multi-colony-stimulating factor or interleukin 3. J Clin Invest 1987; 80: 818–823.
Acknowledgements
These studies were supported by Korea Research Foundation fund (E00026) and Jennerex Biotherapeutics Inc. (San Francisco, CA). We thank Dr Kelley in OHRI (Ottawa) for important comment.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Cancer Gene Therapy website (http://www.nature.com/cgt)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lee, JH., Roh, MS., Lee, YK. et al. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther 17, 73–79 (2010). https://doi.org/10.1038/cgt.2009.50
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2009.50
Keywords
This article is cited by
-
In situ dendritic cell vaccination for the treatment of glioma and literature review
Tumor Biology (2016)
-
A novel recombinant protein of ephrinA1–PE38/GM-CSF activate dendritic cells vaccine in rats with glioma
Tumor Biology (2015)
-
Going viral with cancer immunotherapy
Nature Reviews Cancer (2014)
-
Activation of the human immune system by chemotherapeutic or targeted agents combined with the oncolytic parvovirus H-1
BMC Cancer (2011)
-
Recent advances in oncolytic virus design
Clinical and Translational Oncology (2011)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}