
Abstract
Amyloid-beta (Aβ) is a hallmark component of age-related macular degeneration (AMD), which induces secretion of pro-inflammatory cytokines from retinal pigment epithelium (RPE). Previous studies have shown that p50/RelA (p65), a member of NF-κB family, is an essential pro-inflammatory transcription factor responding to Aβ1-40 stimulation, but few focused on the other two Rel transcription factor members – RelB and c-Rel – and their role in Aβ1-40-mediated inflammation. It was reported that RelA, RelB and c-Rel are also implicated in various NF-κB-mediated inflammatory diseases. Therefore, we infer that Aβ1-40-mediated inflammation targets not only the classical inflammation regulator, RelA, but also RelB and c-Rel. In this study, we demonstrate that intravitreally injected Aβ1-40 mice develop AMD-like pathologic changes, coupled with Rel protein (RelA, RelB and c-Rel) synthesis and nuclear translocation. To focus on the interaction mechanism of Rel proteins, we found that RelB and c-Rel formed a heterodimer with RelA in mice model. We also found that c-Rel silencing decreased the levels of Aβ1-40-dependent RelA expression, indicating that RelB and c-Rel may interact with RelA as coactivator and c-Rel is required to activate the expression of RelA. Moreover, Rel protein silencing decreased the expression of distinct pro-inflammatory cytokines. Together, we demonstrate that besides RelA, RelB and c-Rel can also be activated by Aβ1-40, all of which mediate pro-inflammatory cytokine transcription and RPE damage. Our findings imply that RPE-mediated inflammation under the stimulation of Aβ1-40 is multi-targeted and RelA, RelB and c-Rel proteins may be the new targets of anti-inflammatory agents.
Similar content being viewed by others


Main
Age-related macular degeneration (AMD) is one of the principal causes of visual impairment among aged population in developed countries.1 Therapies targeting vascular endothelial growth factor substantially improve the visual acuity of patients with exudative/wet AMD. Unfortunately, nonexudative/dry AMD, the other subtype of AMD that accounts for up to 90% of cases, has no established treatment.2, 3, 4 It is universally acknowledged that AMD has a multifaceted etiology. Excessive inflammation, including complement activation,5 inflammasome activation6 and accumulation of immune activators, is an important factor in the development of AMD. Despite advances in the understanding of AMD pathogenesis, the underlying mechanism of how inflammation facilitates the development of this disease is still under investigation.
The pathogenesis of early AMD is characterized by subretinal pigment epithelial deposits – drusen, which is composed of a range of proteins and lipids.7 Increasing evidences support that amyloid-beta (Aβ), one of the components of drusen, is correlated with the pro-inflammatory events in retinal pigment epithelial (RPE) cells.8, 9 Compared with other forms of Aβ, Aβ1-40 is the most prevalent type in drusen.10 Both subretinal and intravitreal injections (IVLs) of Aβ upregulate the expression of inflammatory mediators in RPE cells and develop AMD-like pathological changes.9, 11 The mechanism by which Aβ triggers inflammation in the retina is still not fully understood. It was reported that Aβ1-40 deposition triggered inflammation in the retina via activation of the NF-κB pathway. Liu et al. demonstrated that NF-κB/RelA activation was enhanced in RPE cells after the stimulation of Aβ1-40. The NF-κB pathway is suppressed by vinpocetine, resulting in reduced expression levels of IL-1β, IL-18, NLRP3 and other cytokines.12 In addition, Lee et al. proved that Aβ1-40 induced activation of NF-κB/RelA and TNF-α mRNA expression.13
Inducible transcription factors in the NF-κB family exist as homodimers or heterodimers of five distinct proteins, including RelA (p65), RelB and c-Rel (Rel), p50 and p52, which contain a highly conserved N-terminal Rel homology domain. The p50 and p52 proteins are unable to activate transcription independently, as they lack transactivation domains. In contrast, RelA (p65), RelB and c-Rel, defined as the ‘Rel proteins’, have C-terminal transactivation domains.14 NF-κB dimers normally stay inactive in the cytoplasm and transfer into the nucleus when they are activated. Despite the fact that p50 and p52 might have some potential effect on physiopathogenesis of inflammation, we explored a possible role of Rel proteins in Aβ1-40-mediated inflammation in the present study. As the most prototypical NF-κB dimer, the p50/RelA heterodimer is associated with inflammation, oxidative stress and cellular homeostasis in numerous diseases, including AMD. In addition to RelA, the other two members of the Rel family, RelB and c-Rel, are primarily restricted to immune cells. RelB−/− or c-Rel−/− mice develop impaired cellular immunity,15, 16 suggesting their close connection with inflammation. However, the functions of RelB and c-Rel in an Aβ-induced AMD model have not yet been investigated. Therefore, in the current study, we proved that Aβ1-40 can activate RelA, RelB and c-Rel both in vivo and in vitro, all of which co-mediate inflammatory reactions. Our study has a implication that RPE-mediated inflammation under the stimulation of Aβ1-40 is multi-targeted, thus RelA, RelB and c-Rel proteins may be the new targets of anti-inflammatory agents.
Results
IVL of Aβ1-40 induces RPE damage and pro-inflammatory cytokine expression
The penetration of intravitreally injected Aβ1-40 has been proved through rat retinas9, 17 but not through the retinas of C57BL/6 mice. To verify the deposition of Aβ1-40 on RPE cells, we performed immunofluorescence on retinal sections. In contrast to vehicle-injected eyes, increased Aβ signaling was observed at days 2 and 4 after IVL, and it was distributed predominantly in the photoreceptor outer segment and RPE cells (Figure 1a). This finding suggests that Aβ1-40 can spread widely through the mice retina and reach the RPE cells. To assess whether the Aβ1-40 deposition induces RPE cell senescence, transmission electron microscopy (TEM) was used to examine the RPE structures. The RPE cells in the phosphate-buffered saline (PBS)-injected mice appeared normal in morphology, whereas the RPE in the Aβ1-40 mice exhibited damage structures (Figure 1b). Specifically, both the BrM and RPE basal in-foldings were thicker, which were similar to the ultrastructural alterations in APOE4-HFC-induced AMD models, in which the expression of Aβ1-40/42 elevated.18 To test whether or not visual function was impaired by Aβ peptide, electroretinogram (ERG) was performed at day 4 since the injection. Significant reductions in the scotopic a- and b-wave ERG amplitudes in the Aβ1-40-injected mice were detected compared with those in control group, with 29% and 43% average decrease in the a and b waves, respectively (Figure 1c). The reduction in a-wave amplitudes suggests the impairment in photoreceptor and RPE. Next, we isolated RPE–choroid complexes and measured cytokine mRNA expression by qPCR after the injection of Aβ1-40. The expression level of five cytokines significantly increased under the Aβ1-40 stimulation on day 4: IL-1β (2.0±0.1-fold); IL-6 (4.2±0.6-fold); IL-8 (2.0±0.1-fold); IL-18 (2.7±0.1-fold); and IL-12b (6.2±1.5-fold; Figure 1d).

Aβ1-40 induces RPE impairment and upregulates pro-inflammatory cytokine expression in vivo. C57BL/6 mice were injected intravitreally with 5 μl of 2.8 μg/μl Aβ1-40 (oligomeric form) or vehicle (PBS). (a) Immunoreactivity for Aβ1-40 was detected in the OS and RPE cells (arrows) at days 1 and 4 in the Aβ-injected sections. Scale bar: 50 μm. (b) TEM of the RPE and Bruch’s membrane regions in mice. Obvious thickening of the BrM and RPE basal in-foldings with ultrastructural alterations (Scale bar: 5 μm) was observed compared with that in the PBS-injected control mice (Scale bar: 1 μm). (c) Demonstration of waveforms of the maximal ERG response and amplitude evaluations of the scotopic ERG responses were recorded. (d) IL-1β, IL-18, IL-6, IL-8 and IL-12b mRNA expression levels in RPE–choroid in response to Aβ1-40 at day 4. BrM, Bruch’s membrane; CC, choriocapillaris; OS, outer segment. Histograms represent the mean and S.E.M. N=8, NS=nonsignificant P-value, *P<0.05, **P<0.01, ***P<0.001 via Student’s t-tests
Altogether, these data suggest that the IVL of Aβ1-40 resulted in inflammation, which led to AMD-like pathology and induced an impaired visual function in mice.
Rel protein levels increased in the Aβ1-40-mediated AMD model
It is known that RelA is an enhancer of Aβ-induced cytokine transcription in RPE cells; however, RelB and c-Rel are also the key regulators of numerous pro-inflammatory cytokines.19, 20 To investigate whether Aβ1-40 affects Rel protein expression in vivo, Rel mRNA expression in the RPE–choroid complex was evaluated by quantitative real-time RT-PCR. Although there were no significant differences in RelA mRNA levels, the RelB mRNA levels were upregulated (1.81±0.06-fold), and the c-Rel mRNA levels increased (2.30±0.12-fold) at day 2 (Figure 2a). Next, Rel protein expression was assessed in RPE–choroid complex cytoplasm exposed to Aβ1-40 at different time points. Compared with the controls, Aβ1-40 treatment resulted in significant increases in the RelB and c-Rel cytoplasm expression levels at day 2, followed by an elevation in RelA levels at day 4 (Figure 2b). In addition, increased RelB and c-Rel nucleoprotein expression levels were observed at day 2, followed by a constant high expression level until day 4, whereas RelA was upregulated only at day 4. It suggests a different time pattern of Rel protein activation, which is consistent to the Rel protein expression in cytoplasm described above (Figure 2c). We further investigated the potential role of Aβ1-40 in primary mouse RPE cells. The primary mouse RPE cells were measured at 6, 12 and 24 h of culture in the presence of 5 μM Aβ1-40. RelB and c-Rel were significantly upregulated in RPE cells at 12 h. After 24 h, the expression level of c-Rel decreased; however, the RelB level still remained higher than the controls. The RelA level was the same between the treatments and the time before 12 h, but they started to elevate at 24 h (Figure 2d). Our data show that the cytoplasmic and nuclear Rel protein levels were elevated following Aβ1-40 stimulation in a time-dependent pattern.

A time-dependent Rel protein expression pattern was induced by Aβ1-40 both in vivo and in vitro. (a) qRT-PCR assays of Rel gene expression in RPE–choroid complexes. Western blots showing time-dependent expression of Rel proteins in cytoplasmic (b) and nuclear extracts (c) of WT RPE–choroid complexes treated with 5 μl of 2.8 μg/μl Aβ1-40 or PBS (0, 2 and 4 days). Histone H3 was used as a quality control for the nuclear extracts. (d) Rel protein expression in mouse RPE cells in response to 5 μM Aβ1-40 at 6, 12 and 24 h after treatment. UT, untreated; WT, wild type. Histograms represent the mean and S.E.M. N=3, NS=nonsignificant P-value, *P<0.05, **P<0.01, ***P<0.001 via Student’s t-tests
Aβ1-40 triggers Rel nuclear translocation and activation
To figure out the effect of the Rel proteins on the activation of pro-inflammatory cytokine transcription, we detected alterations in Rel protein distribution in response to Aβ1-40 stimulation. Initially, we confirmed the nuclear translocation of the Rel proteins at the different time points as described above (Figures 2b and c).
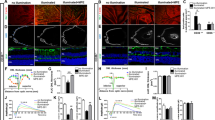
Next, we used confocal microscopic analysis to detect Rel protein nuclear translocation. RelB and c-Rel showed strong fluorescence signal in the nuclei of RPE cells, which were marked by RPE65 at day 4, whereas no nuclear signaling was observed in the PBS group (Figure 3a). For further confirmation, primary mouse RPE cells were incubated with Aβ1-40 for 24 h. Nuclear Rel protein expression was hardly observed in the PBS group; however, the nuclear fluorescence signal significantly increased in the Aβ1-40 group (Figure 3b). These results indicate that Rel protein translocation into the nucleus can be triggered by Aβ1-40 in a periodic manner.

Rel protein translocation in RPE–choroid complexes and in primary mouse RPE cells. (a) Retinal sections co-immunostained with RPE65 (green) and RelB/c-Rel(red) at day 4. RPE nuclei were marked by white arrows (Scale bar: 20 μm). (b) Confocal microscopic analysis showing the distribution of the RelA/RelB/c-Rel signal (red) at 24 h after 5 μM Aβ1-40 or PBS treatment. Nuclei were immunostained with DAPI (blue) (Scale bar: 50 μm)
Aβ1-40 promotes c-Rel-dependent expression of RelA and the formation of RelA–RelB/c-Rel complexes
To assess whether RelB and c-Rel were associated with the expression of RelA, siRNAs targeting RelB and c-Rel were transfected into ARPE-19 cells to silence their expression. After 48 h of siRNA treatment, the ARPE-19 cells were stimulated with Aβ1-40. As shown in Figure 4a, c-Rel silencing decreased the Aβ1-40-dependent RelA expression level, which was similar to those achieved by RelA silencing alone. In contrast, RelA silencing had no effect on either RelB or c-Rel expression, regardless of exposure to Aβ1-40 (Figure 4a). In addition, RelB or c-Rel overexpression by transfection with their respective lentivirus activation particles led to a increase in the other two Rel protein members in primary mouse RPE cells (Figure 4b). These results suggest that there is a crosstalk and a common pathway among the Rel proteins and c-Rel in specific positively regulates the expression of RelA after Aβ1-40 stimulation in RPE cells.

The interaction between Rel proteins in RPE–choroid complex and in RPE cells. (a) RelA, RelB and c-Rel silencing induced changes in the RelA, RelB and c-Rel expression levels in ARPE-19 cells, as determined by qRT-PCR analysis. (b) qRT-PCR assays of Rel mRNA expression induced by RelA, RelB and c-Rel overexpression in primary mouse RPE cells. (c) Co-immunoprecipitation of c-Rel, RelB and RelA at 4 days post injection of Aβ1-40 or PBS. Histograms represent the mean and S.E.M. N=3, NS=nonsignificant P-value, *P<0.05, **P<0.01 via Student’s t-tests
Furthermore, RelA may be an activator as part of the RelA–RelB/c-Rel dimer. Therefore, RelA in the RPE–choroid complex was co-immunoprecipitated with RelB and c-Rel at day 4 after injection. The data strongly indicate that RelB and c-Rel formed heterodimers with RelA after IVL of Aβ1-40 (Figure 4c). In summary, these results suggest that RelA activity might be affected by the preferential nuclear translocation of the dimers RelA–RelB and RelA–c-Rel, and the expression of c-Rel in Aβ1-40-stimulated RPE cells.
Rel proteins are the intracellular messengers of Aβ1-40-mediated inflammation
To confirm whether RelB/c-Rel are key mediators of pro-inflammatory cytokines, as RelA is, we silenced their expression in ARPE-19 cells. Consistent with the above in vivo results, IL-1β, IL-8, IL-18, IL-6 and IL-12 mRNA levels were upregulated in ARPE-19 cells 24 h after exposure to Aβ1-40. The statistical analysis showed the significant differences in Rel protein expression levels following siRNA treatment compared with those in control siRNA. The IL-1β, IL-18 and IL-12b in the Aβ1-40 group decreased to a similar level in RelA, RelB and c-Rel knocked-down RPE cells in contrast to control siRNA, whereas IL-6 and IL-8 mRNA were downregulated in the RelB- and c-Rel-silenced groups to a greater degree than RelA, despite partial inhibition in the RelA-silenced group (Figures 5a–e). Moreover, Aβ1-40-induced IL-6 expression significantly decreased only in the c-Rel-silenced RPE cells (Figure 5a), and Aβ1-40-induced IL-12b expression significantly decreased only in the RelB-silenced RPE cells (Figure 5e). These results indicate that although RelA is central to these processes in Aβ-mediated RPE inflammation, RelB and c-Rel silencing also decreases cytokine expression to a greater extent, suggesting a different RelB/c-Rel-dependent mode of regulation. The c-Rel-mediated regulation of IL-1β, IL-18 and IL-12b expression may depend on RelA according to the results above, which needs further investigation. The schematic diagram of Rel proteins in pathogenesis of Aβ1-40-mediated inflammation is illustrated in Figure 5f.

Effects of Rel protein silencing on pro-inflammatory cytokine levels in RPE cells. (a–f) ARPE-19 cells were transfected with only Lipofectamine 6000 reagent, non-siRNA, RelA siRNA, RelB siRNA or c-Rel siRNA for 24 h. IL-6 (a), IL-1β (b), IL-8 (c), IL-18 (d) and IL-12b (e) mRNA levels were evaluated by qRT-PCR. The mRNA levels were standardized to housekeeping gene expression. (f) Schematization of the model obtained by the integration of the data obtained in this report and the context. Histograms represent the mean and S.E.M. N=3, NS=nonsignificant P-value, *P<0.05, **P<0.01 Student’s t-tests
Discussion
In this study we demonstrate that Aβ1-40 activates the pro-inflammatory transcription factor Rel proteins in vivo and in RPE cells. The present data also show that not only RelA but also RelB and c-Rel are activated, and RelB and c-Rel form heterodimers with RelA in a Aβ1-40-related AMD model. Our study established for the first time that the effects of Aβ1-40 in RPE-mediated inflammation are multi-targeted.
First, we proved that intravitreally injected Aβ1-40 reaches RPE cells, which results in an impairment of the retina. Our prior study showed that RPE cells from Aβ1-40-injected mice displayed hyper- and hypopigmentation.21 In the current study, the structure and function of the retina were evaluated by TEM and ERG, respectively. The ultrastructural alterations and decreased ERG responses observed in our study were similar to those reported in other AMD animal models.22, 23 However, these studies investigated only the effects of certain risk factors in AMD development, such as pathologic changes. As there are no generally established AMD animal models we focused on the influence of Aβ1-40 on RPE cells in this study. The model we used may partially explain the association between Aβ1-40 and retinal degeneration in AMD.
Our previous study showed that RelA and the IκB kinase inhibitor of κB kinase epsilon, which are essential in regulating NF-κB-mediated inflammatory responses, were implicated in informatics analyses of chip results in an Aβ1-40-injected model.21 Subsequent to our prior results, the current study provided evidence that Aβ activates transcription factor Rel proteins in a time-dependent pattern in vivo and in RPE cells. Given that RelA has been well studied in Aβ1-40-mediated inflammation, we focused on the biological functions of RelB and c-Rel in this study. We demonstrated that Aβ1-40 induces increased translocation of RelB and c-Rel into the nucleus before RelA, whose expression may be regulated by c-Rel (Figures 2b and c). The time-dependent pattern of Rel protein expression has also been reported in other studies,24, 25 which leads to the hypothesis that RelB and c-Rel, along with RelA, might have important roles in different time periods;26 however, this possibility requires further investigation. In addition, the association of RelA with RelB and c-Rel was markedly increased in response to Aβ1-40 stimulation in RPE cells (Figure 5c). Previous reports have suggested that distinct combinations of Rel proteins generate specific NF-κB-responsive gene profiles by affecting transcriptional activation, although the Rel dimer composition depends on the cell type and cellular stimuli. Jacque and colleagues described inhibition of RelA DNA binding by RelA–RelB complex formation after lipopolysaccharide (LPS) stimulation in lymphoid cells.27 RelA–c-Rel dimers initiated transcriptional amplification of c-Rel and the formation of c-Rel–c-Rel dimers, which exert protective effects on RPE cells.28 Therefore, it is likely that RelB and c-Rel may affect cytokine expression through the following mechanisms: (a) modulation of RelA activity by direct complex formation; (b) modulation of RelA expression; and (c) directly binding to cytokine promotors without RelA in Aβ1-40-stimulated RPE cells.
We have shown that Rel proteins enhance pro-inflammatory cytokine secretion in RPE cells. There is a strong link between these cytokines and AMD in patients.29, 30 Maturation of IL-1β and its analog, IL-18, was promoted by NLRP3 inflammasome activation, a pathway essential in both dry and wet AMD.31 IL-1β and IL-18 have cytotoxic effects in inducing RPE degeneration. In addition, IL-1β promotes neovascularization,32 whereas contrasting findings have been reported that IL-18 inhibits angiogenesis in choroidal neovascularization.33 Thus, whether exposure to IL-1β and IL-18 results in AMD-like pathology remains an important question. IL-6 and IL-8 are prominent cytokines that mediate chronic inflammatory responses shared across a variety of age-related pathologies and are implicated in AMD progression following LPS stimulation.34, 35 IL-6 also had a pro-angiogenic effect in an AMD model, similar to IL-1β and IL-18.36 IL-12b upregulation may also be a risk factor in AMD.37 These cytokines can enhance the synthesis of other cytokines, suggesting that the Aβ1-40 toxicity mediated by these cytokines through an autocrine feedback loop may impair RPE cells and stimulate angiogenesis, contributing to AMD pathogenesis. Among them, IL-1β, IL-18 and IL-12b secretion levels were impaired in RelA- or c-Rel/RelB-silenced RPE cells, whereas the IL-6 and IL-8 mRNA levels were diminished only in the c-Rel- or RelB-deleted RPE cells, but not in the RelA-knockdown RPE cells. These two different regulation patterns by the Rel proteins on these two cytokine expression profiles may reveal their different functions in AMD, although their underlying mechanisms remain unknown. However, conflicting results have also been reported that RelB recruitment to target genes is associated with transcriptional downregulation of IL-12b, while our data indicated that the IL-12b mRNA level was significantly decreased in the RelB-silenced group (Figure 5e). These controversial results are perhaps due to the different roles of RelB in distinct cells or stimuli.38 DNA binding of Rel proteins to cytokine promoters requires further exploration. We infer that therapy targeting RelB/c-Rel may be a more effective strategy to reduce Aβ1-40-mediated RPE damage.
In summary, our data confirm the following hypotheses: (1) activation of RelA, RelB and c-Rel occurs in Aβ1-40-stimulated RPE cells; (2) activated Rel proteins positively regulate inflammation; and (3) inflammation is alleviated in RPE cells as long as one of the Rel proteins is knocked out. Three Rel proteins may now be considered important regulators of Aβ1-40-induced RPE degeneration. These findings indicate the multi-targeted effects of Aβ1-40, which shed light on endogenous signaling in Aβ-induced AMD. Hopefully, it can provide novel therapeutic approaches for RPE protection in AMD.
Materials and methods
Amyloid oligomerization
Aβ1-40 oligomeric peptide (GL Biochemistry, Shanghai, China) was prepared as previously described.39 Briefly, synthetic lyophilized Aβ1-40 peptides were dissolved in deionized distilled water at a concentration of 28 μg/μl. Aβ solutions were diluted to reach a final concentration of 2.8 μg/μl by immediate addition of PBS and incubated for 7 days at 37 °C. The samples were stored at −80 °C for use. Electron microscopy was used to verify the aggregated states of Aβ1-40.
Animal model and treatment
Two-month-old male C57BL/6 mice were supplied by the Laboratory Animal Center at the Shanghai First People’s Hospital. The animals were reared and housed in sterilized enclosures with a 12 h light cycle.
The mice were anesthetized with 1.5% sodium pentobarbital (100 μl/20 g intraperitoneally (i.p.)) and were administered a single unilateral IVL of Aβ1-40 peptides (14 μg/5 μl) in PBS using a glass micropipette to deliver oligomeric Aβ1-40 peptides under a dissecting microscope (SM2000J; Olympus, Tokyo, Japan). The Aβ1-40 peptide concentration was reported in a previous study.21 Briefly, age-matched controls received 5 μl of PBS in the same manner (n=8). The mice eyes were enucleated under i.p. anesthesia after 2 and 4 days since the injection. All animal experiments were approved by the Ethics Committee of Jiao Tong University, Shanghai, China, and were conducted in compliance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Primary mouse RPE cell isolation and culture
To isolate RPE cells from 3-week-old wild-type C57BL/6 mice for primary culture, the anterior portion of the eye and retina were removed gently with forceps, and then 0.25% trypsin (Gibco, Carlsbad, CA, USA) was added to the eyecups for 20 min at 37 °C in a 5% CO2 atmosphere. After trypsin treatment, the RPE sheets were peeled off under a dissecting microscope. The collected single-cell suspension was transferred to complete Dulbecco’s modified Eagle’s medium /Ham’s F-12 medium (Gibco) supplemented with 10% fetal bovine serum, 1% non-essential amino acids and 1% HEPES (Gibco). The cells were passed every 3 days by 0.25% trypsin (Gibco).
Electroretinography
Scotopic ERG was performed using RETIport System (Roland Consult, Brandenburg, Germany) with a Super Color Ganzfeld (Q450 SC) stimulator. The animals were dark-adapted overnight, and then were anesthetized with an i.p. of 1% sodium pentobarbital under red illumination. Pupils were dilated with atropine (0.5%). Contact lens electrodes were placed on the cornea following topical anesthesia of the cornea. The ground electrode was placed midway up the tail, and then the scotopic ERG was recorded. All manipulations were done under scotopic conditions. The amplitude of the a wave/b wave, defined as the scope from the baseline to the respective trough, was measured and analyzed using a built-in software.
Real-time quantitative PCR
Total RNA extraction and quantification were performed according to the RNAsimple Total Kit protocol (Tiangen Biotech, Beijing, China). NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) was used to quantify the RNA samples. Following RNA extraction, RT Master Mix (Takara Bio Inc., Dalian, China) was used to generate cDNA with according to the manufacturer’s protocol.
The primers were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize all samples. The specific sense and antisense primer sequences used are available Table 1. The RT-PCRs by a SYBR green-based PCR method were performed using a real-time PCR detection system (Eppendorf, Hamburg, Germany) with a program of 40 cycles of amplification (95 °C for 5 s, 60 °C for 30 s and 72 °C for 42 s). The relative expression level of each mRNA was calculated using the 2−ΔΔCt method.
Western blot analysis
RPE–choroid tissues or RPE cells were collected and lysed with radio-immunoprecipitation assay buffer containing proteinase inhibitors. Nuclear and cytoplasmic protein were extracted using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China). Aliquots of each sample were subjected to 10% SDS-PAGE gels and electroblotted to polyvinylidene difluoride membranes (Merck Millipore, Billerica, MA, USA). The membranes were blocked by blocking buffer (Tris-buffered saline Tween-20 (TBST), containing 5% nonfat dry milk) for 1 h at room temperature and incubated with primary antibodies against RelA (1:1000, Cell Signaling Technology, Beverly, MA, USA), RelB (1:2000, Abcam, Cambridge, MA, USA), c-Rel (1:2000, Abcam), GAPDH (1:1000, Cell Signaling Technology) or Histone H3 (1:1000, Cell Signaling Technology) overnight. The membranes were then washed with TBST three times, and then probed with horseradish peroxidase-conjugated secondary antibodies (1:2000, Proteintech, Chicago, IL, USA) for 1 h at room temperature. The membranes were washed with TBST and then were exposed to a molecular imaging system (Amersham Imager 600, GE Healthcare, Buckinghamshire, UK).
Immunocytochemistry and TEM
Immunocytochemistry assays were performed on retinal sections or in 24-well slide chambers. Briefly, after fixation, the samples were blocked with 0.3% Triton X-100 and 5% goat serum albumin (Beyotime) in PBS for 1 h at room temperature. Tissue sections were immunostained with primary antibodies against RelA (1:300, Cell Signaling Technology), RelB (1:300, Abcam), c-Rel (1:300, Abcam) and RPE65 (1:300, Novus Biologicals, Littleton, CO, USA) overnight at 4 °C. The samples were washed with PBS three times and then were stained for 45 min at 37 °C with Alexa Fluor 594- and 488-conjugated secondary antibodies (1:1 000, Proteintech). The nuclei were marked by 4′,6-diamidino-2-phenylindole. RPE cells were visualized using a fluorescence microscope (Olympus) and a Leica TCS SP8 confocal laser scanning microscope (Leica TCS NT, Wetzlar, Germany).
For ultrastructural analysis, the samples were send to Shanghai FuDan University School of Medicine after fixation in 2.5% glutaraldehyde (dissolved in PBS) at 4 °C. The ultrathin sections were observed under an electron microscope (Tecnai G2 spirit twin, FEI, Eindhoven, The Netherlands).
Lentiviral activation particles
Primary RPE cells were transduced with lentiviral-mediated RelB-specific CRISPR/dCas9 activation plasmid (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Activated clones were selected via puromycin dihydrochloride (Santa Cruz Biotechnology) administration at 4 days after the transduction. RNA was isolated for activation analysis by RT-PCR 2 days later.
RNA interference
ARPE-19 cells were transfected with double-stranded siRNA or negative control siRNA (non-siRNA) using Lipo6000 Transfection Reagent (Beyotime). Target sequences were as follows: 5′-GCCCUAUCCCUUUACGUCA-3′ for p65, 5′-GAGGACAUAUCAGUGGUGUUCAGCA-3′ for RelB and 5′-AAAUGUGAAGGGCGAUCAGCA-3′ for c-Rel. Double-stranded siRNAs were synthesized by Shanghai GenePharma (Shanghai, China).
Co-immunoprecipitation and immunoblotting
Co-immunoprecipitation studies were performed on RPE–choroid complexes according to the Pierce Co-Immunoprecipitation Kit (Rockford, IL, USA) protocol. The following antibodies, rabbit anti-RelA (Cell Signaling Technology), rabbit anti-RelB/c-Rel (Abcam) or rabbit IgG (Santa Cruz Biotechnology), were used for immunoprecipitation. The immunoprecipitation products were analyzed via western blot. The following antibodies, mouse anti-RelA (Cell Signaling Technology) or mouse anti-Relb/c-Rel (Santa Cruz Biotechnology), were used for western blot analysis.
Statistical analysis
Each experiment was repeated in triplicate. The data were expressed as the mean±S.E.M., if applicable. Two-tailed unpaired Student’s t-tests (GraphPad Prism, San Diego, CA, USA) were used to calculate the P-value of the results. A P-value <0.05 was considered to be statistically significant.
References
Klein R, Peto T, Bird A, Vannewkirk MR . The epidemiology of age-related macular degeneration. Am J Ophthalmol 2004; 137: 486–495.
Group C R, Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011; 364: 1897–1908.
Investigators I S, Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ et al. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology 2012; 119: 1399–1411.
Chew EY, Clemons TE, Agron E, Sperduto RD, Sangiovanni JP, Kurinij N et al. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology 2013; 120: 1604–1611 e4.
Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai AK, Ripke S et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 2011; 43: 1232–1236.
Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D'Amore PA et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci 2013; 54: 110–120.
Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA 2002; 99: 14682–14687.
Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH . The Alzheimer's A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA 2002; 99: 11830–11835.
Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL et al. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Vis Sci 2013; 54: 2225–2237.
Isas JM, Luibl V, Johnson LV, Kayed R, Wetzel R, Glabe CG et al. Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthalmol Vis Sci 2010; 51: 1304–1310.
Liu C, Cao L, Yang S, Xu L, Liu P, Wang F et al. Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Int J Mol Med 2015; 35: 169–176.
Liu RT, Wang A, To E, Gao J, Cao S, Cui JZ et al. Vinpocetine inhibits amyloid-beta induced activation of NF-kappaB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp Eye Res 2014; 127: 49–58.
Lee JJ, Wang PW, Yang IH, Wu CL, Chuang JH . Amyloid-beta mediates the receptor of advanced glycation end product-induced pro-inflammatory response via toll-like receptor 4 signaling pathway in retinal ganglion cell line RGC-5. Int J Biochem Cell Biol 2015; 64: 1–10.
Gilmore TD, Wolenski FS . NF-kappaB: where did it come from and why? Immunol Rev 2012; 246: 14–35.
Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell 1995; 80: 331–340.
Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 1995; 9: 1965–1977.
Howlett DR, Bate ST, Collier S, Lawman A, Chapman T, Ashmeade T et al. Characterisation of amyloid-induced inflammatory responses in the rat retina. Exp Brain Res 2011; 214: 185–197.
Ding JD, Johnson LV, Herrmann R, Farsiu S, Smith SG, Groelle M et al. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci USA 2011; 108: E279–E287.
Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nat Immunol 2012; 13: 1162–1170.
Ren S, Zhang S, Li M, Huang C, Liang R, Jiang A et al. NF-kappaB p65 and c-Rel subunits promote phagocytosis and cytokine secretion by splenic macrophages in cirrhotic patients with hypersplenism. Int J Biochem Cell Biol 2013; 45: 335–343.
Huang P, Sun J, Wang F, Luo X, Feng J, Gu Q et al. MicroRNA expression patterns involved in amyloid beta-induced retinal degeneration. Invest Ophthalmol Vis Sci 2017; 58: 1726–1735.
Ufret-Vincenty RL, Aredo B, Liu X, McMahon A, Chen PW, Sun H et al. Transgenic mice expressing variants of complement factor H develop AMD-like retinal findings. Invest Ophthalmol Vis Sci 2010; 51: 5878–5887.
Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW . Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci 2006; 47: 729–737.
Hu YC, Sun Q, Li W, Zhang DD, Ma B, Li S et al. Biphasic activation of nuclear factor kappa B and expression of p65 and c-Rel after traumatic brain injury in rats. Inflamm Res 2014; 63: 109–115.
You WC, Li W, Zhuang Z, Tang Y, Lu HC, Ji XJ et al. Biphasic activation of nuclear factor-kappa B in experimental models of subarachnoid hemorrhage in vivo and in vitro. Mediators Inflamm 2012; 2012: 786242.
Hoffmann A, Levchenko A, Scott ML, Baltimore D . The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 2002; 298: 1241–1245.
Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M . RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem 2003; 278: 19852–19860.
Calandria JM, Asatryan A, Balaszczuk V, Knott EJ, Jun BK, Mukherjee PK et al. NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival. Cell Death Differ 2015; 22: 1363–1377.
Tsai YY, Lin JM, Wan L, Lin HJ, Tsai Y, Lee CC et al. Interleukin gene polymorphisms in age-related macular degeneration. Invest Ophthalmol Vis Sci 2008; 49: 693–698.
Cao S, Ko A, Partanen M, Pakzad-Vaezi K, Merkur AB, Albiani DA et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am J Ophthalmol 2013; 156: 1176–1183.
Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012; 149: 847–859.
Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B et al. Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol 2011; 178: 2416–2423.
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 2012; 18: 791–798.
Leung KW, Barnstable CJ, Tombran-Tink J . Bacterial endotoxin activates retinal pigment epithelial cells and induces their degeneration through IL-6 and IL-8 autocrine signaling. Mol Immunol 2009; 46: 1374–1386.
van Deursen JM . The role of senescent cells in ageing. Nature 2014; 509: 439–446.
Izumi-Nagai K, Nagai N, Ozawa Y, Mihara M, Ohsugi Y, Kurihara T et al. Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. Am J Pathol 2007; 170: 2149–2158.
Chen M, Forrester JV, Xu H . Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS ONE 2011; 6: e22818.
Saccani S, Pantano S, Natoli G . Modulation of NF-kappaB activity by exchange of dimers. Mol Cell 2003; 11: 1563–1574.
Xing S, Shen D, Chen C, Wang J, Liu T, Yu Z . Regulation of neuronal toxicity of beta-amyloid oligomers by surface ATP synthase. Mol Med Rep 2013; 8: 1689–1694.
Acknowledgements
The study was supported by the National Science Fund for Distinguished Young Scholars (81425006), Translational Medicine Innovation Fund of Shanghai Jiao Tong University School of Medicine (15ZH4005), Science and Technology Commission of Shanghai Municipality (16dz2251500 and 16140900800) and Program for Eastern Young Scholar at Shanghai Institutions of Higher Learning (QD2016003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Publisher’s Note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, J., Huang, P., Liang, J. et al. Cooperation of Rel family members in regulating Aβ1-40-mediated pro-inflammatory cytokine secretion by retinal pigment epithelial cells. Cell Death Dis 8, e3115 (2017). https://doi.org/10.1038/cddis.2017.502
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.502
This article is cited by
-
Immunoproteasome Subunit Low Molecular Mass Peptide 2 (LMP2) Deficiency Ameliorates LPS/Aβ1-42-Induced Neuroinflammation
Molecular Neurobiology (2024)
-
The retinal toxicity profile towards assemblies of Amyloid-β indicate the predominant pathophysiological activity of oligomeric species
Scientific Reports (2020)
-
Effects of concentration of amyloid β (Aβ) on viability of cultured retinal pigment epithelial cells
BMC Ophthalmology (2019)
