
Abstract
The long noncoding RNAs (lncRNAs) have long been clarified to participate in hepatocellular carcinoma (HCC) as a biomarker. We carried out the present study in order to identify HCC-related lncRNAs and elucidate the functional roles in the development and progression of HCC. Our previous study has provided that LINC01225 may be an HCC-related gene. Here, we verified that LINC01225 was upregulated in HCC. Knockdown of LINC01225 resulted in inhibited cell proliferation and invasion with activated apoptosis and cell cycle arrest in vitro. Overexpression of LINC01225 in LINC01225 knockdown cells presented that attenuated cell proliferation and invasion were restored and enhanced. Subcutaneous and tail vein/intraperitoneal injection xenotransplantation model in vivo validated reduced tumor progression and metastasis. Investigation of mechanism found that LINC01225 could bind to epidermal growth factor receptor (EGFR) and increase the protein level of EGFR, and subsequently fine tune the EGFR/Ras/Raf-1/MEK/MAPK signaling pathway. Analysis with clinicopathological information suggested a high expression of LINC01225 is positively associated with poor prognosis. We also proved that LINC01225 was stably expressed in serum and can act as a novel biomarker in predicting the diagnosis of HCC. As a conclusion, LINC01225 plays a crucial role in HCC and can act as a biomarker for the diagnosis and prognosis of HCC.
Similar content being viewed by others


Main
Hepatocellular carcinoma (HCC) accounts for 70–90% of primary liver cancer, which has emerged as the fifth most common cancer and the second leading cause of cancer-related death worldwide.1, 2 Tumor biomarkers may serve as an early indicator of cancer.3, 4, 5 However, few biomarkers have been identified associated with the genesis, growth and metastasis of HCC. It is of paramount importance to elucidate the relationship between clinical symptoms and molecular changes in HCC in order to identify new strategies for diagnosis and treatment and improve prognosis.
Long noncoding RNAs (lncRNAs) range from 200 nt up to ~100 kb in length. During the past decade, several studies have implicated that lncRNAs play pivotal roles in physiological and pathological processes.6 Deregulated lncRNAs were identified in HCC; several of which were confirmed as biomarkers for predicting survival and metastasis of HCC.7, 8, 9, 10 However, the function and clinical significance of most lncRNAs in the development of HCC remain largely unknown. Transcriptomic sequencing and bioinformatics analyses are widely used to detect lncRNAs within the human genome and identify putative candidate genes involved in carcinogenesis. Based on such methods, we found that an lncRNA, LINC01225, was deregulated in HCC. Investigations of its function and mechanism in combination with clinical information analysis consistently suggested that LINC01225 is a potential diagnostic biomarker and therapeutic target for HCC.
Results
High expression of LINC01225 in HCC
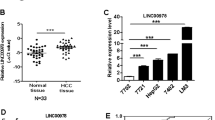
In our previous study, microarray detection and bioinformatics showed that expression of LINC01225 (mentioned as LOC149086) was increased in serum from patients with HCC.11 In consideration of the detection bias attributed to the limited samples provided, a large hospital-based case–control study was performed. As the result of real-time PCR showed, the LINC01225 expression level in serum from patients with HCC was elevated in comparison with serum from healthy controls, which verified the result of microarray analysis (Figure 1a). This suggested that LINC01225 was abnormally expressed in HCC. To study the relationship between LINC01225 and HCC, we detected expression of LINC01225 in 180 pairs of HCC tumors compared with the corresponding adjacent tissues by real-time PCR, which suggested that LINC01225 transcript levels were higher in tumor tissues compared with non-tumor tissues (Figure 1b). Moreover, expression of LINC01225 was detected in several HCC cell lines, including SMCC7721, MHCC97H, HepG2 and Huh7 and normal liver cell line, L02 (Figure 1c). The result showed that LINC01225 is unregulated in some HCC cell lines. In addition, we detected the location of LINC01225 transcript by real-time PCR amplified with separated nuclear and cytoplasm RNA and found LINC01225 was located primarily in the cytoplasm of MHCC97H cells (Figure 1d).

Aberrant upregulation of LINC01225 in HCC; subcellular location. (a) Increased level of LINC01225 in HCC serum (n=66) was detected in comparison with serum from healthy controls (n=70). The relative expression of LINC01225 versus Has-5s, calculated by 2−△△CT, was log transformed and presented as box plots. Box plot explanation: upper horizontal line of box, 75th percentile; lower horizontal line of box, 25th percentile; horizontal bar within box, median; upper horizontal bar outside box, 95th percentile; lower horizontal bar outside box, 5th percentile. (b) Increased level of LINC01225 was detected in HCC tissues compared with the corresponding adjacent tissues (n=180). The relative expression of LINC01225 versus Has-5 s, calculated by 2−△△CT, was log transformed and presented as box plots. Box plot explanation: upper horizontal line of box, 75th percentile; lower horizontal line of box, 25th percentile; horizontal bar within box, median; upper horizontal bar outside box, 95th percentile; lower horizontal bar outside box, 5th percentile. (c) Level of LINC01225 was detected in HCC cell lines and L02 cells. Data were 2△△CT transformed and presented as mean±S.E.M. (d) Subcellular localization indicated that the transcript for LINC01225 was located primarily in the cytoplasm of MHCC97H cells, according to quantitative real-time PCR amplification with separated cytoplasmic and nuclear RNA. U2 was used as the control for cytonuclear expression and HPRT for cytoplasmic expression. Data were log transformed and presented as mean±S.E.M. Human Has-5 s was used as control. (**P<0.01, ***P<0.001)
The analysis for clinicopathological information of 180 HCC patients showed that high LINC01225 expression was significantly associated with tumor size, tumor differentiation grade, tumor capsular integrity, tumor TNM stage and metastasis (Table 1). Meanwhile, the analysis with HCC patients’ plasma also suggested that high LINC01225 expression was significantly associated with tumor size, tumor differentiation grade, tumor capsular integrity, tumor TNM stage and metastasis (Table 2).
LINC01225 promotes proliferation and invasion of HCC cells in vitro
In order to detect the function of LINC01225 in regulating cell biological behavior, SMCC7721 and MHCC97H were selected as high-expression LINC01225 cells. shRNA1 and shRNA2 plasmid constructed with lentivirus plasmid, named Lv-shRNA and Lv-shRNA2 respectively, was used to knockdown the expression of LINC01225 and further applied to detect the biological significance of LINC01225 in tumor growth and metastasis (Supplementary Figure S1A and B). The results of EdU assay (Figure 2a) and CCK8 assay (Figure 2b) indicated that the reduced expression of LINC01225 significantly inhibited the proliferation of SMCC7721 and MHCC97H cells. Overexpression of LINC01225 in LINC01225 knockdown cells presented that attenuated cell proliferation was restored and enhanced.

LINC01225 promoted cell proliferation and invasion in vitro. (a) Edu assay confirmed the functional role of LINC01225 in cell proliferation (× 200). (b) The results of Edu assay. Decreased level of LINC01225 reduced the proliferation of SMCC7721 and MHCC97H after 24 h, compared with the control cells; overexpression of LINC01225 in LINC01225 knockdown cells restored and enhanced cell proliferation after 24 h. The integral optical density (IOD) values of cells treated with control plasmids were normalized to 100%. (c) Upper panels: CCK8 assay showed that LINC01225 stable knockdown inhibited growth of SMCC7721 and MHCC97H cells; cell growth was restored and enhanced in LINC01225 knockdown cells treated with Lv-Rescue plasmid. Lower panel: Absorbance at 450 nm was presented as the mean±S.E.M. Absorbance at 450 nm of cells treated with control plasmids was normalized to 100%. Data were collected and provided at 24 h after cultivation. (d) Left panels: Morphology of invasive SMCC7721 and MHCC97H cells after stable transfection with negative control, LINC01225 shRNA or Lv-Rescue plasmid (× 400). Right panel: The number of cells treated with control plasmid was normalized to 100%, and data are presented as the mean±S.E.M., based on at least three independent experiments (*P<0.05, **P<0.01, ***P<0.01)
The Transwell assay was conducted to determine the functional role of LINC01225 in cell invasion. Cell migration was significantly reduced in SMCC7721 and MHCC97H cells treated with shRNA in contrast to control cells; cell migration was restored and enhanced in cells treated with Lv-Rescue plasmid (Figure 2c).
FACS technology was applied to determine the influence of LINC01225 on apoptosis. The results suggested that cells with decreased expression of LINC01225 had a higher level of apoptosis (Figure 3a). The detection of the role of LINC01225 in the regulation of cell cycle showed that treatment with shRNA resulted in observable cell cycle arrest (Figure 3b). Overexpression of LINC01225 in LINC01225 knockdown cells have restored the cell ability with low level of apoptosis and reduced cell cycle arrest (Figures 3a and b). Accordingly, we speculated that LINC01225 could promote cell proliferation and invasion.

Knockdown of LINC01225 resulted in activated apoptosis and cell cycle arrest. (a) Left panels: Forty-eight hours after treatment with negative control, LINC01225 shRNA or Lv-Rescue plasmid, cells were stained and analyzed by flow cytometry. LR, early apoptotic cells; UR, terminal apoptotic cells. Right panels: Ratio of early apoptotic cells was collected and presented in the column chart. (b) Upper panels: At 48 h after treatment with negative control, LINC01225 shRNA or Lv-Rescue plasmid, cell cycle was analyzed by flow cytometry. Lower panels: The bar chart represents the percentage of cells in the G1, S or G2 phase. All experiments were performed in triplicate and presented as the mean±S.E.M. (*P<0.05, **P<0.01, ***P<0.001)
LINC01225 acted as a promoter of tumor growth and metastasis in vivo
A xenotransplantation model was used to determine the role of LINC01225 in tumorigenesis. Compared with the control group, tumors in mice injected with SMCC7721 and MHCC97H cells transfected with Lv-shRNA were significantly smaller (Figure 4a). Hematoxylin and eosin (H&E) staining of the tumors is shown in Figure 4a. Detection of abdominal metastasis in a nude mouse model transplanted intraperitoneally with LINC01225 stably knocked down cells or control cells indicated that decreased expression of LINC01225 was significantly related to reduced invasion and abdominal metastases of SMCC7721 and MHCC97H cells in accordance with photon flux detection (Figure 4b). Lung metastases formed in 7/10 SMCC7721 control group mice and 9/10 MHCC97H control group mice, and 1/10 SMCC7721-Lv-shRNA group mice and 2/10 MHCC97H-Lv-shRNA mice (Figure 4c). These results were verified by histological examination.

LINC01225 downregulation inhibited tumor growth and metastasis due to LINC01225 knockdown in vivo. (a) Upper panels: The right groin of BALB/C nude mice was subcutaneously transplanted with SMCC7721 or MHCC97H cells stably transfected with LINC01225 shRNA or control plasmids (n=5). As indicated by arrows, 4 weeks after implantation, LINC01225 depletion attenuated tumor growth in nude mice. Lower panels: The weight of each tumor was measured. Data are presented as the mean±S.E.M. H&E staining of the tumors is shown (× 400). (b) Left panels: Tumor model of intraperitoneal transplantation with LINC01225 stably knocked down cells or control cells in BALB/C nude mice was constructed to detect abdominal metastases. Fluorescence intensity was measured using the IVIS Lumina II system. Mice injected with SMCC7721 and MHCC97H cells stably knocked down with LINC01225 suppressed metastasis of tumor cells compared with controls. Right panel: Fluorescence intensity in control cells was normalized to 100%; data were presented as the mean±S.E.M. (c) Left panels: Representative figures of tail vein xenograft model indicated lung colonization, which was formed in SMCC7721 and MHCC97H cells (n=10). Right panel: The number of mice with pulmonary metastastic foci was calculated in each group, as presented in the table. Lung colonization was formed in 7/10 SMCC7721 control group mice and 9/10 MHCC97H control group mice; 1/10 of the LINC01225 stably knocked down SMCC7721 group and 2/10 of the LINC01225 stably knocked down MHCC97H group also had lung colonization. The χ2 test was used to analyze the difference. H&E staining of the lung tissues is shown. (× 400). All experiments were performed in triplicate and presented as the mean±S.E.M. (**P<0.01, ***P<0.001)
LINC01225 promotes HCC growth and invasion via EGFR/MAPK pathway
Microarray was performed with LINC01225 stable knockdown cells and mock empty plasmid of pll3.7-treated cells. As shown in Supplementary Figure S1C, 4/0.25 was set as the cutoff values to select candidate aberrant expression genes for gene set enrichment analysis. As indicated by gene-annotation enrichment analysis, the mitogen-activated protein kinase (MAPK) signaling pathway had a high correlation with downregulation of LINC01225 (Supplementary Figure S1D).
Mass spectrometry (MS) analysis suggested that EGFR was decreased due to LINC01225 depletion (Figure 5a), which was in accordance with the result of microarray analysis (data not shown). Thus, we selected EGFR as a target of LINC01225, through which it might regulate the MAPK signaling pathway. The different expression of EGFR was confirmed by both quantitative real-time PCR, western blotting and enzyme-linked immunosorbent assay (ELISA) assay (Figure 5b and Supplementary Figure S1E). We found that there was more significant change at the protein level than at the mRNA level. Additionally, the LINC01225 transcript was located primarily in the cytoplasm of MHCC97H cells, as indicated by the results of real-time PCR amplified with separated nuclear and cytoplasm RNA (Figure 1d), which demonstrated that LINC01225 may interact with proteins in the cytoplasm, rather than acting as a novel transcription factor. We therefore hypothesized that regulation of EGFR by LINC01225 at the protein level is more important than at the mRNA level. To verify this assumption, we carried out RNA immunoprecipitation (RIP) (Figure 5c), which showed that LINC01225 could bind to EGFR, thereby increase the protein level of EGFR.

LINC01225 promotes HCC growth and invasion via the EGFR/MAPK pathway. (a) Upper panel: Polyacrylamide gel stained with rapid silver staining showed that EGFR was reduced in LINC01225 knocked down MHCC97H cells. Lower panel: MS analysis was performed to discover abnormally expressed protein due to LINC01225 depletion. Trypsin digestion fragment detected by MS was presented in the panel. Other fragments detected were VLGSGAFGTVYK, YSFGATcVK, cNLLEGEPR, EISDGDVIISGNK, ELIIEFSK, ELVEPLTPSGEAPNQALLR, ESDcLVcR, GDSFTHTPPLDPQELDILK and RPAGSVQNPVYHNQPLNPAPSR. b and y stands for N-terminal and C-terminal collision-induced dissociation fragment ions. (b) Upper panel: the EGFR protein expression level was detected by western blotting. Lower panel: ELISA was used to detect the protein of EGFR in cell lysate. Cells were treated with control plasmid, Lv-shRNA, Lv-shRNA2 and Lv-Rescue plasmid. (c) Upper panel: RIP was performed using an antibody against EGFR on extracts from MHCC97H cells with IgG as a negative control and MHCC97H cells treated with EGFR-shRNA as an additional control. Enrichment of LINC01225 was normalized to the input. Purified RNA was used for real-time PCR analysis. Lower panel: Bands were detected from the RNA in the group with anti-EGFR, and attenuated bands were detected in LINC01225 knockdown cells. (d) Expression of proteins participating in the EGFR/MAPK pathway detected by western blotting. Protein level of Ras, activated Raf-1 and MEK was reduced by LINC01225 depletion. (e) p44/42 and activated p-p44/42 protein expression level was detected by western blotting. The protein level of activated p44/42 was decreased when LINC01225 was knocked down. All experiments were performed in triplicate and presented as the mean±S.E.M. (*P<0.05, ***P<0.001)
Western blotting indicated that the level of Ras and activated Raf-1 was reduced, along with activated MEK (mitogen-activated protein kinase/p44/42 kinase) and MAPK, phosphorylated p44/42 (Figures 5d and e). These findings demonstrated that LINC01225 has a modifying effect on HCC via reducing the activation of the EGFR/Ras/Raf-1/MEK/MAPK pathway.
LINC01225 as a biomarker for HCC diagnosis and prognosis
Previous studies have reported lncRNAs as biomarkers for diagnosis and predictors for prognosis of HCC.7, 8, 9, 10 We performed Kaplan–Meier analysis to identify whether expression of LINC01225 was associated with the cancer-specific survival rate in HCC patients. With the median as the cutoff based on the expression level of LINC01225 in HCC tissues, patients were separated into a high-level group (n=90) and a low-level group (n=90). Expression of LINC01225 was negatively associated with cancer-specific survival (P<0.01) (Figure 6a). We performed risk score analysis to investigate the effectiveness of LINC01225 for HCC prediction. Due to the limitations that serum samples corresponding to the HCC tissues could not be obtained, we therefore randomly selected several HCC serum samples (n=66) collected from patients before hepatectomy and control serum samples (n=70) were collected from people undergoing physical examination. Serum samples were frozen and thawed for five times, and expression of LINC01225 in serum detected by qRT-PCR indicated that LINC01225 was stably expressed in serum (Supplementary Figure S1F). LINC01225 may be an effective predictor for HCC diagnosis with a sensitivity of 0.761 and a specificity of 0.443 (Figure 6b).

LINC01225 as a biomarker for HCC diagnosis and prognosis. (a) Kaplan–Meier analysis was initiated to discover whether the expression of LINC01225 was associated with overall survival of HCC patients. The median was used as the cutoff. (b) Expression of LINC01225 was detected in patients from whom plasma was obtained preoperatively, by comparison with patients free of tumor. Receiver operating characteristic curve analysis of LINC01225 was used to detect the diagnostic efficiency of HCC.
Discussion
HCC is one of the most common cancers and has a high mortality.12 HCC is rarely detected in the early stages because of its slow progression and early lack of symptoms. This means that patients are mostly diagnosed at an advanced stage and cannot be cured.13, 14 Clinical research has revealed that HCC detected at the early stages has good prognosis and high overall survival rate after treatment with transhepatic arterial chemotherapy and embolization or radiotherapy.2, 15, 16 On account of the significance of early detection of HCC, several methods were developed, such as traditional diagnostic imaging with ultrasound, computed tomography and diagnostic biomarkers. Tumor biomarkers are specifically expressed in some diseases or differentially expressed between patients and healthy individuals, and can be detected in body fluids or secretions. Biomarkers may be used as indicators of tumorigenesis and for surveillance of tumor progression and metastasis, as well as serving as predictors of prognosis and survival. In addition to metabolites and tumor-specific antigens, tumor-related genes have emerged as promising biomarkers with high specificity and sensitivity.17, 18, 19
lncRNAs regulate a wide variety of physiological and pathological processes through diverse mechanisms; however, previously, they were thought to be transcriptional noise.20, 21 Accumulating studies have demonstrated that lncRNAs have important functions and are associated with a wide range of diseases.22, 23, 24, 25 lncRNAs are involved in all aspects of gene regulation, including chromosome dosage compensation, imprinting, epigenetic regulation, nuclear and cytoplasmic trafficking, transcription, and mRNA splicing and translation.7, 26, 27 HCC is a complex polygenetic disease with many coding and noncoding genes involved.28, 29 Many studies have suggested that several lncRNAs play important roles in the development and progression of HCC, such as HOTAIR, HULC, MALAT1 and H19.30, 31, 32, 33 Due to the undiscovered functions of lncRNAs and limited knowledge of the underlying mechanisms, more studies are urgently needed. The present study found an HCC-related lncRNA, LINC01225, and elucidated its functional roles in HCC development and progression.
As described in the NCBI database, LINC01225 is a human noncoding RNA validated by the Human Gene Nomenclature Committee. It is located at chromosome 1p35.2 with 2113 bp and has eight exons. Our previous study reported that LINC01225 was highly expressed in serum of patients with HCC, which suggested that LINC01225 is an HCC-related gene.11 In the present study, we found that LINC01225 was highly expressed in serum and tumor tissues of patients with HCC. High expression of LINC01225 was positively related to tumor size and metastasis, as suggested by the analysis of clinicopathological information. Our in vitro experiment demonstrated that the absence of LINC01225 was responsible for reduced cell proliferation and invasiveness in HCC, and re-expression of LINC01225 in LINC01225 knockdown cells showed that attenuated cell proliferation and cell migration were restored and enhanced. Moreover, cell cycle arrest was obviously increased and apoptosis was markedly promoted by LINC01225 depletion and re-expression of LINC01225 in LINC01225 knockdown cells has restored the cell ability with a low level of apoptosis and reduced cell cycle arrest. Our in vivo study of the effect of LINC01225 showed that reduced tumor size and depressed abdominal metastasis were attributed to depletion of LINC01225. Lung metastatic node formation was decreased by LINC01225 knockdown, which further suggested that LINC01225 might act as a promoter of HCC.
EGFR plays a pivotal role in cell survival and proliferation.34 Consequent transformation has been observed in model systems of autocrine stimulation, overexpression and mutation of EGFR.35, 36 EGFR exerts its effect by activating a network of signaling elements, involving Ras, phosphoinositide 3-kinase and the signal transducer and activator of transcription family.37, 38 A cascade of protein kinases, with activated phosphorylation by growth factors, is well established, including tyrosine kinases, Ras, Raf-1, MEK and MAPK.39, 40, 41 The Ras/Raf-1/MEK/MAPK signaling pathway is of major importance in intracellular transduction of proliferative signals from activated cell membrane growth factor receptors to the nucleus, and it is involved in cell proliferation and apoptosis in several tumors including HCC.42 Raf-1 is a serine/threonine kinase that is activated by GTP-bound Ras, the activated type of Ras, subsequently phosphorylating downstream signal MEK, which phosphorylates and activates MAPK.43, 44 MAPK phosphorylates a series of cellular substrates with crucial roles in cell survival and proliferation. Whether lncRNAs modify HCC development and progression through activation of these molecules remains vague.
Previous studies have identified lncRNAs as transcriptional co-regulators, through which lncRNAs realized the regulatory roles by interacting with proteins.45, 46, 47, 48 Further investigation of the exact mechanism through which LINC01225 participates in HCC is urgently needed. Microassay combined with MS had determined EGFR as a target of LINC01225. We also found as a result of LINC01225 knockdown that EGFR was dramatically reduced more at the level of protein than mRNA, which suggests that LINC01225 affects EGFR signaling primarily through regulating EGFR protein. GO and KEGG pathway analysis suggested that LINC01225 functions as a regulator by affecting the MAPK signaling pathway.
According to clinicopathological analysis of information at follow-up, high expression of LINC01225 is positively associated with poor prognosis. In addition, we have demonstrated LINC01225 as a novel biomarker in predicting the diagnosis of HCC by analyzing its expression in serum.
In summary, our study shows the upregulation of LINC01225 in HCC, and further demonstrates that LINC01225 plays a crucial role in HCC through binding to protein and increasing the level of EGFR as a consequence, thus fine tuning the EGFR/MAPK signaling pathway. LINC01225 may be a promising biomarker for the diagnosis and prognosis of HCC, and it can be a therapeutic target for HCC. However, more studies are warranted for further investigation of the function of LINC01225 in the future.
Materials and Methods
Patients and clinical samples
Fresh HCC specimens, paired adjuvant noncancerous tissue samples and HCC serum samples were obtained from patients undergoing hepatectomy between 2008 and 2010. Serum samples from healthy persons were randomly selected at the First Affiliated Hospital of Nanjing Medical University (Nanjing, China). The study was approved by our Institutional Ethics Committee. Our research was performed in accordance with the Helsinki Declaration and government policies. Written informed consent was obtained from all participants. Peripheral blood samples were collected before hepatectomy. Specimens and corresponding clinical and pathological materials are summarized in Table 1 (tissues) and Table 2 (serum).
Cell lines and animals
HCC cell lines SMCC7721 and MHCC97H and common human liver cell line L02 were purchased from American Type Culture Collection, and MHCC97H and Huh7 cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, Shanghai, China). The cell lines were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Grand Island, NY, USA) with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA), 2 mM l-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin, in 5% CO2 at 37 °C.
Male BALB/c nu/nu mice (4–8 weeks old) were purchased from the Laboratory Animal Center of Yangzhou University and maintained under specific pathogen-free conditions. All animals received humane care and all experiments were carried out according to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals.
Cell transfection
Two shRNAs of LINC01225 sequences were designed, shRNA1: GGAUAAUUCUAAUGCCUACTTGUAGGCAUUAGAAUUAUCCTT; shRNA2: UCCAGUUGGCGAGAGUGAUTTAUCACUCUCGCCAACUGGATT. SMCC7721 and MHCC97H cells were transfected with lentivirus plasmid constructed with shRNA of LINC01225 as previously described,49 and finally shRNA1 plasmid termed lentivirus-short hairpin RNA (Lv-shRNA) was selected to be used in the subsequent experiments. Lentivirus plasmid constructed with scramble sequence was used as control, termed Lv-NC. The synthesized and purified LINC01225 gene fragment was inserted into a lentivirus vector (pll3.7), named Lv-Rescue. LINC01225 knockdown SMCC7721 and MHCC97H cells were then transfected with the packaged recombinant lentivirus. shRNA of EGFR was designed for the target sequence: GGCTGGTTATGTCCTCATT. Lentivirus plasmid constructed with shRNA of EGFR, termed EGFR-shRNA, was used to transform MHCC97H cells as previously described49 (Supplementary Figure S2).
Quantitative real-time PCR
Quantitative real-time PCR was performed to detect the expression levels of LINC01225 and mRNAs of all related genes with SYBR Green Mastermix kit (TaKaRa, Tokyo, Japan) and analyzed in triplicate assays on the ABI Prism 7900HT (Applied Biosystems, Foster City, CA, USA) according to the direction of the reagents. Total RNAs of fresh liver samples and cells were extracted with TRIzol reagent (Invitrogen) and quantified. For RNA extraction from the cytoplast and cytoplasm, the SurePrep Nuclear or Cytoplasmic RNA Purification Kit (Life Science SOURCE; Biovision, Milpitas, CA, USA) was used. For mRNA detection, reverse transcriptase (TaKaRa) was used to reverse transcribe total RNAs (500 ng). Has-5 S was used as an internal control. Primer sequences: Has-5 S: Forward: 5′-GGAGAGGGAGCCTGAGAAACG-3′ and Reverse: 5′-TTACAGGGCCTCGAAAGAGTCC-3′ and human LINC01225: Forward: 5′-GTCCCTTACCTTGAGGTGCC-3′ and Reverse: 5′-CACGCCTTTGTGTTCTGGTG-3′ and human EGFR: Forward: 5′-TCCTCTGGAGGCTGAGAAAA-3′ and Reverse: 5′-GGGCTCTGGAGGAAAAGAAA-3′ were designed using Primer 3.0 software (http://www.simgene.com/Primer3).
Western blotting and H&E staining and ELISA
Total proteins from cultured cells were extracted using radio-immunoprecipitation assay buffer plus fresh protease and phosphatase inhibitors (Beyotime, Nantong, China) and quantified with the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein samples (30 μg) were loaded to each lane. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride membrane. Antibodies against EGFR (Cell Signaling Technology, Danvers, MA, USA), Ras (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Raf-1 (Santa Cruz Biotechnology), p-Raf-1 (Thr269) (Santa Cruz Biotechnology), MEK (Cell Signaling Technology), phosphorylated MEK (p-MEK; Cell Signaling Technology), p44/42 (ERK) (Cell Signaling Technology), p-p44/42 (Thr202/Tyr204) (Cell Signaling Technology) and human reduced glyceraldehyde-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology) were used in immunoblotting. ImageJ software (NIH, Bethesda, MD, USA) was applied to quantify the integrated density of the bands. Cells suffering freezing and freeze–thaw cycles were used to extract protein for ELISA assay, which was performed with an ELISA Kit for EGFR (Uscn Life Science Inc., Wuhan, China) according to the instruction.
Flow cytometry analysis
Apoptosis was detected with an Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme Biotech, Nanjing, China) and cell cycle was determined with a Cell cycle Assay Kit (Vazyme Biotech). Cells used for apoptosis detection were treated with 0.05 mM H2O2 for 2 h for stimulation of apoptosis. Cells were finally analyzed on a FACS Calibur flow cytometer equipped with CellQuest software (BD Biosciences, New York, NY, USA).
Cell proliferation and invasion assay
Cell proliferation was evaluated using a CCK8 kit (Vazyme Biotech) and a Cell-Light EdU Apollo567 In Vitro Kit (RiboBio, Guangzhou, China). For CCK8 detection, transfected cells (2 × 103) were seeded into 96-well plates, cultured for 24, 48, 72 and 96 h, and then CCK8 reagent was added to each well and incubated for 2 h at 37 °C. Absorption was measured by microplate reader at 450 nm (ELX-800; Bio-Tek, Winooski, VT, USA). For 5-ethynyl-2′-deoxyuridine (EdU) assay, transfected cells (2 × 105) were seeded into Glass Botttom Cell Culture Dishes (Nest Biotechnology, NJ, USA), then cells were treated according manufacture instruction, and finally cell samples were detected with a laser confocal scanning microscopy. The invasion assays were assessed using the Transwell units (Corning Costar, Tewksbury, MA, USA) percolated with Matrigel (BD Biosciences). Cells (2 × 104 cells/well) were seeded at the upper compartment of the chamber in DMEM without FBS, and the lower chamber was filled with DMEM including 10% FBS as chemotaxin. After incubation for 48 h, the filters were collected, fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Non-invading tumor cells on the top of the filters were removed with cotton swabs. Cells passing through the filter were counted under a light microscopy.
Subcutaneous xenotransplantation model
Cells (5 × 106) with a stably decreased level of LINC01225 expression in SMCC7721 and MHCC97H and control cells (Lv-NC) were subcutaneously implanted into the bilateral axillae of each BALB/C nude mice. Five weeks later, all mice were killed and the weight of each tumor was measured. Tumor tissues were integrally stripped out.
Metastasis model
A single-cell suspension was prepared with the cells stably knocked down for LINC01225, or in the control group, suspended in 200 μl PBS and filtered through a sterile 70-μm nylon mesh filter (BD Falcon, Franklin Lakes, NJ, USA). Groups of 10 mice were separately injected with SMCC7721 and MHCC97H cells with or without knockdown of LINC01225 via tail vein injection to develop peripheral intravascular implanted models (10 in each group). Mice were killed 5 weeks afterwards, and tumor metastasis in the lung was examined. H&E staining was used to evaluate the number of tumors in lung tissues.
In addition, a tumor model of intraperitoneal transplantation in BALB/C nude mice was constructed. Mice were injected intraperitoneally with cells treated with LINC01225 shRNA or controls and kept in pathogen-free conditions. Abdominal metastasis was monitored weekly by the IVIS Lumina II system (Caliper Life Sciences, Hopkinton, MA, USA).
Agarose gel electrophoresis
Agarose powder (0.5 g) was dissolved in 50 ml of 0.5% Tris-acetate-EDTA (TAE) buffer and heated to near boiling, and then 2.5 μl GoldView (Beyotime) was added and mixed. Total RNA of the cell lines was extracted and reverse transcribed into cDNAs. Obtained cDNAs were mixed with loading buffer (Beyotime), and the mixture was loaded into the wells. Electrophoresis was performed with 0.5% TAE as the running buffer, at 80 V for 40 min. Data were analyzed with Image Lab software performed with an ultraviolet (UV) transilluminator.
Microarray assay detection and bioinformatics analysis
Total RNAs extracted from MHCC97H cells including untreated cells, LINC01225 stable knockdown cells and mock empty plasmid of PLL3.7-treated cells were amplified and used to synthesize double-stranded cDNAs. Samples prepared as required were labeled and hybridized to mRNA Human Gene Expression Microarray V4.0 (CapitalBio Corp., Beijing, China). The microarray results were used to analyze data normalization, summarization and quality control with the GeneSpring software V11.5 (Agilent, Santa Clara, CA, USA). We selected genes that were differentially expressed with a >2-fold change of threshold value, as well as Benjamini–Hochberg corrected P-values of <0.05. CLUSTER version 3.0 software (Stanford University School of Medicine, Stanford, CA, USA) was used to normalize and hierarchically cluster the data. Tree visualization of the data was performed with Java Treeview software (Stanford University School of Medicine, Stanford, CA, USA). Microarray data have been deposited at ArrayExpress (E-MTAB-3833).
Gene product attributes were described with three structured networks of defined terms provided with GO analysis from Gene Ontology (www.geneontology.org). The GO Term enrichment with expressed mRNA list was considered significantly different with P<0.05. Pathway analysis was performed on the basis of the latest KEGG (Kyoto Encyclopedia of Genes and Genomes) database for the differentially expressed mRNAs with a statistically significant P-value of <0.05.
Mass spectrometry
Polyacrylamide gel was prepared in accordance with the standard protocol. Twenty microliters of each sample mixed with 10 × running buffer (CapitalBio Corp.) was loaded, and the gel was run at 120 V for 2.5 h. The gel was stained with Fast Silver Stain Kit (Beyotime). Lanes, cut into 10 pieces were respectively placed in Eppendorf tubes. NH4HCO3 (50 mM) was added and the gel was broken into pieces with the suction pipette. Then, trypsin (Promega, Fitchburg, WI, USA) was used to digest the gel solution at 37 °C overnight. Tubes were shaken and centrifuged with methyl cyanide (HPLC grade) added for 2 min at room temperature. The supernatant was transferred to the clean tubes, and dried by the vacuum concentration apparatus at 60 °C. LC-MS/MS with nano-LC combined with an Orbitrap Q Exactive mass spectrometer (Thermo Scientific, Waltham, MA, USA) was used for analysis of the peptides followed by a scan range of m/z 400–1500. Thermo Proteome Discoverer (1.4.0.288) software (http://www.thermoscientific.com/en/product/proteome-discoverer-software.html) platform was used to analyze the raw files. MS/MS spectra were retrieved against the protein database (human-refseq-20140303-71465 s.fasta, National Center of Biotechnology Information) for protein authentication.
RNA immunoprecipitation
RIP was performed with a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA, USA). An antibody for EGFR (Cell Signaling Technology) was used for RIP assays. Coprecipitated RNAs were detected using quantitative real-time PCR.
Statistical analysis
Results of quantitative real-time PCR were expressed as mean±S.E.M. Student’s t-test and χ2 test were used to evaluate statistical differences in clinical and demographic characteristics. Kaplan–Meier survival curves were drawn and the log-rank test was performed. Risk score analysis was done to investigate the effectiveness of the LINC01225 for prediction. Statistical analysis was performed using STATA version 9.2 (Stata Corp., College Station, TX, USA) and SPSS version 18.0 (SPSS Inc., Chicago, IL, USA) and presented with the GraphPad prism software (GraphPad Software, San Diego, CA, USA). In all cases, P<0.05 was considered significant.
Abbreviations
- HCC:
-
hepatocellular carcinoma
- lncRNA:
-
long noncoding RNA
- EGFR:
-
epidermal growth factor receptor
- MEK:
-
mitogen-activated protein kinase/p44/42 kinase
- ERK:
-
extracellular signal-regulated kinase
- MAPK:
-
mitogen-activated protein kinase
- RIP:
-
RNA immunoprecipitation
- shRNAs:
-
short hairpin RNAs
- Lv-shRNA:
-
lentivirus-short hairpin RNA
- GAPDH:
-
glyceraldehyde-phosphate dehydrogenase
- ELISA:
-
enzyme-linked immunosorbent assay
- IOD:
-
integral optical density
References
Shimakawa Y, Lemoine M, Njai HF, Bottomley C, Ndow G, Goldin RD et al. Natural history of chronic HBV infection in West Africa: a longitudinal population-based study from The Gambia. Gut 2015 (e-pub ahead of print; doi: 10.1136/gutjnl-2015-309892.
Maluccio M, Covey A . Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin 2012; 62: 394–399.
Hou J, Zhou Y, Zheng Y, Fan J, Zhou W, Ng IO et al. Hepatic RIG-I predicts survival and interferon-alpha therapeutic response in hepatocellular carcinoma. Cancer Cell 2014; 25: 49–63.
Kucuk O . Cancer biomarkers. Mol Aspects Med 2015; 45: 1–2.
Quagliata L, Matter MS, Piscuoglio S, Arabi L, Ruiz C, Procino A et al. Long noncoding RNA HOTTIP/HOXA13 expression is associated with disease progression and predicts outcome in hepatocellular carcinoma patients. Hepatology 2014; 59: 911–923.
Wahlestedt C . Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov 2013; 12: 433–446.
Yang X, Xie X, Xiao YF, Xie R, Hu CJ, Tang B et al. The emergence of long non-coding RNAs in the tumorigenesis of hepatocellular carcinoma. Cancer Lett 2015; 360: 119–124.
Suh B, Park S, Shin DW, Yun JM, Yang HK, Yu SJ et al. High liver fibrosis index FIB-4 is highly predictive of hepatocellular carcinoma in chronic hepatitis B carriers. Hepatology 2015; 61: 1261–1268.
Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z et al. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene 2015 (e-pub ahead of print; doi: 10.1038/onc.2015.223.
Tang J, Zhuo H, Zhang X, Jiang R, Ji J, Deng L et al. A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis 2014; 5: e1549.
Tang J, Jiang R, Deng L, Zhang X, Wang K, Sun B . Circulation long non-coding RNAs act as biomarkers for predicting tumorigenesis and metastasis in hepatocellular carcinoma. Oncotarget 2015; 6: 4505–4515.
Liu LX, Lee NP, Chan VW, Xue W, Zender L, Zhang C et al. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology 2009; 50: 1453–1463.
Aravalli RN, Steer CJ, Cressman EN . Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008; 48: 2047–2063.
Goyal L, Muzumdar MD, Zhu AX . Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res 2013; 19: 2310–2318.
El-Serag HB, Marrero JA, Rudolph L, Reddy KR . Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008; 134: 1752–1763.
Venook AP . Treatment of hepatocellular carcinoma: too many options? J Clin Oncol 1994; 12: 1323–1334.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21: 938–945.
Li J, Tan S, Kooger R, Zhang C, Zhang Y . MicroRNAs as novel biological targets for detection and regulation. Chem Soc Rev 2014; 43: 506–517.
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 2015; 25: 981–984.
Fatica A, Bozzoni I . Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 2014; 15: 7–21.
Flintoft L . Non-coding RNA: ribosomes, but no translation, for lincRNAs. Nat Rev Genet 2013; 14: 520.
Yang Y, Cai Y, Wu G, Chen X, Liu Y, Wang X et al. Plasma long non-coding RNA, CoroMarker, a novel biomarker for diagnosis of coronary artery disease. Clin Sci (Lond) 2015; 129: 675–685.
Zhou J, Yu L, Gao X, Hu J, Wang J, Dai Z et al. Plasma microRNA panel to diagnose hepatitis B virus-related hepatocellular carcinoma. J Clin Oncol 2011; 29: 4781–4788.
Zhuang J, Lu Q, Shen B, Huang X, Shen L, Zheng X et al. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep 2015; 5: 11924.
Mehler MF, Mattick JS . Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev 2007; 87: 799–823.
Sun T, Wong N . Transforming growth factor-beta-induced long noncoding RNA promotes liver cancer metastasis via RNA-RNA crosstalk. Hepatology 2015; 61: 722–724.
Takahashi K, Yan I, Haga H, Patel T . Long noncoding RNA in liver diseases. Hepatology 2014; 60: 744–753.
Bard-Chapeau EA, Nguyen AT, Rust AG, Sayadi A, Lee P, Chua BQ et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet 2014; 46: 24–32.
Shukla R, Upton KR, Munoz-Lopez M, Gerhardt DJ, Fisher ME, Nguyen T et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 2013; 153: 101–111.
Fu WM, Zhu X, Wang WM, Lu YF, Hu BG, Wang H et al. Hotair mediates hepatocarcinogenesis through suppressing MiRNA-218 expression and activating P14 and P16 signaling. J Hepatol 2015; 63: 886–895.
Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY, Zhang F et al. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol 2012; 29: 1810–1816.
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007; 132: 330–342.
Zhang L, Yang F, Yuan JH, Yuan SX, Zhou WP, Huo XS et al. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 2013; 34: 577–586.
Kannan S, De Santis M, Lohmeyer M, Riese DJ II, Smith GH, Hynes N et al. Cripto enhances the tyrosine phosphorylation of Shc and activates mitogen-activated protein kinase (MAPK) in mammary epithelial cells. J Biol Chem 1997; 272: 3330–3335.
Garrett TP, Burgess AW, Gan HK, Luwor RB, Cartwright G, Walker F et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc Natl Acad Sci USA 2009; 106: 5082–5087.
Greenall SA, Donoghue JF, Gottardo NG, Johns TG, Adams TE . Glioma-specific Domain IV EGFR cysteine mutations promote ligand-induced covalent receptor dimerization and display enhanced sensitivity to dacomitinib in vivo. Oncogene 2015; 34: 1658–1666.
Huang CW, Tsai HL, Chen YT, Huang CM, Ma CJ, Lu CY et al. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer 2013; 13: 599.
Luo SF, Wang CC, Chiu CT, Chien CS, Hsiao LD, Lin CH et al. Lipopolysaccharide enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Br J Pharmacol 2000; 130: 1799–1808.
Lin CC, Shyr MH, Chien CS, Wang CC, Chiu CT, Hsiao LD et al. Thrombin-stimulated cell proliferation mediated through activation of Ras/Raf/MEK/MAPK pathway in canine cultured tracheal smooth muscle cells. Cell Signal 2002; 14: 265–275.
Tu MT, Luo SF, Wang CC, Chien CS, Chiu CT, Lin CC et al. P2Y(2) receptor-mediated proliferation of C(6) glioma cells via activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 2000; 129: 1481–1489.
Yang CM, Chien CS, Hsiao LD, Pan SL, Wang CC, Chiu CT et al. Mitogenic effect of oxidized low-density lipoprotein on vascular smooth muscle cells mediated by activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 2001; 132: 1531–1541.
Hsu YM, Chiu CT, Wang CC, Chien CS, Luo SF, Hsiao LD et al. Tumour necrosis factor-alpha enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Cell Signal 2001; 13: 633–643.
Bachleitner-Hofmann T, Sun MY, Chen CT, Liska D, Zeng Z, Viale A et al. Antitumor activity of SNX-2112, a synthetic heat shock protein-90 inhibitor, in MET-amplified tumor cells with or without resistance to selective MET Inhibition. Clin Cancer Res 2011; 17: 122–133.
Troiani T, Napolitano S, Vitagliano D, Morgillo F, Capasso A, Sforza V et al. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res 2014; 20: 3775–3786.
Zhu Y, Rowley MJ, Bohmdorfer G, Wierzbicki AT . A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol Cell 2013; 49: 298–309.
Quinodoz S, Guttman M . Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol 2014; 24: 651–663.
McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015; 521: 232–236.
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477: 295–300.
Xu D, Wang X, Yan S, Yin Y, Hou J, Wang X et al. Interaction of PTPRO and TLR4 signaling in hepatocellular carcinoma. Tumour Biol 2014; 35: 10267–10273.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation for Distinguished Young Scholars (Grant no. 81225017 to BS), National Basic Research Program of China (Grant no. 2012CB910800 to BS) and National Natural Science Foundation (Grant no. 81201528 to RJ). This work was also supported in part by the program for Development of Innovative Research Team in the First Affiliated Hospital of Nanjing Medical University and the Priority Academic Program of Jiangsu Higher Education Institutions. BS is a Yangtze River scholars Distinguished Professor.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Calin
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, X., Zhang, W., Tang, J. et al. LINC01225 promotes occurrence and metastasis of hepatocellular carcinoma in an epidermal growth factor receptor-dependent pathway. Cell Death Dis 7, e2130 (2016). https://doi.org/10.1038/cddis.2016.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.26
This article is cited by
-
Autophagy and gastrointestinal cancers: the behind the scenes role of long non-coding RNAs in initiation, progression, and treatment resistance
Cancer Gene Therapy (2021)
-
The role of long noncoding RNAs in hepatocellular carcinoma
Molecular Cancer (2020)
-
VersicanV1 promotes proliferation and metastasis of hepatocellular carcinoma through the activation of EGFR–PI3K–AKT pathway
Oncogene (2020)
-
Optimizing Human Epidermal Growth Factor for its Endurance and Specificity Via Directed Evolution: Functional Importance of Leucine at Position 8
International Journal of Peptide Research and Therapeutics (2020)
-
circRNA_104075 stimulates YAP-dependent tumorigenesis through the regulation of HNF4a and may serve as a diagnostic marker in hepatocellular carcinoma
Cell Death & Disease (2018)

{kind=link}
{kind=link}